|
|
Post by djoser-xyyman on Jan 30, 2019 11:59:07 GMT -5
|
|
|
|
Post by Tukuler al~Takruri on Jan 30, 2019 13:19:03 GMT -5
Can you allign K's via t/pgm?
My guess is, that algo's hard coded.
Remember, eM's ADMIXTUREs and my reduxes?
I think your K's from top down came out
1 Raspberry
2 Purple
3 Royal
4 Navy
5 Blue
6 Lime
7 Green
8 Yellow
9 Orange
0 Red
Sorting for relationships Top down I get
1 Royal (local isolate; gave <4% to Red and Navy)
2 Navy (local w/4 <3% infusions; gave 20% to Red)
3 Orange (local isolate; majority of grp w/a 3 way infusion)
4 Red (local w/Navy minority; ~10% substrata of Green-Blue grp)
5 Green (local w/Red & Navy substrata; up to 15% of Orange grp)
6 Blue (local w/Purple & Red substrata; forms a Green-Blue grp)
7 Yellow (local w/Purple-Raspberry minor substrata; forms a grp w/Purple)
8 Purple (local w/Yellow substrate; forms a grp w/Yellow)
9 Raspberry (forms a grp w/Purple&Lime; contributes to nearly every pop)
0 Lime (local w/Purple substrate; minority element in a grp w/Purple&Raspberry)
Looking at the redux, in general
1 - who do Royal, Navy, or Orange rep?
2 - what region does the GreenBlueRed grp rep?
3 - what region does the YellowPurple grp rep?
|
|
|
|
Post by djoser-xyyman on Jan 30, 2019 14:24:10 GMT -5
============
My comparision datasets fron downlaods from different formats. All need to be reformated back to VCF, merged and reconverted to PLINK bed bim fam. This sdatset goes back to 2012 includes Sahara Tauregs, San. Xhosa Yemeni etc
plink bed to ped
plink --bfile filename --recode --tab --out myfavpedfile
plink --bfile hgdp--mySubset --recode --tab --out hgdp--mySubset
plink --bfile yemen --recode --tab --out yemen
plink --bfile NW-Africa-Qatar-txt-set --recode --tab --out NW-Africa-Qatar-txt-set
plink --bfile northafrica_syria_filtered --recode --tab --out northafrica_syria_filtered
NW-Africa-Qatar-txt-set
yemen
northafrica_syria_filtered
hgdp--mySubset
=============================
----
ped file to VCF
plink --ped file.ped --map file.map --recode vcf --out ped_to_vcf
plink --file your_ped_map_input --recode vcf
plink --ped NW-Africa-Qatar-txt-set.ped --map NW-Africa-Qatar-txt-set.map --recode vcf --out NW-Africa-Qatar-txt-set
plink --ped yemen.ped --map yemen.map --recode vcf --out yemen
plink --ped northafrica_syria_filtered.ped --map northafrica_syria_filtered.map --recode vcf --out northafrica_syria_filtered
plink --ped NW-Africa-Qatar-txt-set.ped --map NW-Africa-Qatar-txt-set.map --recode vcf --out NW-Africa-Qatar-txt-set
datafiles
NW-Africa-Qatar-txt-set
yemen
northafrica_syria_filtered
hgdp--mySubset
===============
|
|
|
|
Post by djoser-xyyman on Jan 30, 2019 14:42:17 GMT -5
@ Sage I am putting a tutorial together. But right now I post pieces as I complete bits and pieces. It may be helpful to some people if they are stuck.
Right now I am concentrating on format conversion because there is a lot of dataset out there going back to 2012. The alignment is a problem so I am working through those issues of conversion making sure alignment is done properly.
Running ADMIXTURE I have gotten. Now, I am making sure I am choosing and inputting the correct datasets. As many Sahara and East SSA and southern Arabia populations like Yemen with Scandinavians/Finns and Papuans being outliers.
.
|
|
|
|
Post by Tukuler al~Takruri on Jan 30, 2019 16:16:11 GMT -5
It's OK I figured it out. Top down by the redux | 1 | Royal | Oceania | | 2 | Navy | Americas | | 3 | Orange | East Asia | | 4 | Red | Central/South Asia | | 5 | Green | Europe | | 6 | Blue | Southwest Asia | | 7 | Yellow | East Africa | | 8 | Purple | West & Central African | | 9 | Raspberry | Northeast Africa | | 10 | Lime | 'Basal Africans |
Yes you got it down. Data in was clean. Yr ADMX run conforms to expectations. @ Sage I am putting a tutorial together. But right now I post pieces as I complete bits and pieces. It may be helpful to some people if they are stuck. Right now I am concentrating on format conversion because there is a lot of dataset out there going back to 2012. The alignment is a problem so I am working through those issues of conversion making sure alignment is done properly. Running ADMIXTURE I have gotten. Now, I am making sure I am choosing and inputting the correct datasets. As many Sahara and East SSA and southern Arabia populations like Yemen with Scandinavians/Finns and Papuans being outliers. . |
|
|
|
Post by djoser-xyyman on Jan 31, 2019 16:03:10 GMT -5
Yeah! We have ADMIXTURE down to some extent but we need to create our own datasets. That means making sure all the popualtions in the dataset is align with the same hg19 or h18 or GrCh38 reference. Conversion back and forth must be done correctly conversion tools needed ---------- I may have to use PlinkLiftOver/LiftOver for my datasets prior to GrCh38 especially those obtained prior to 2013...like the Henn Qatari dataset? On Linux this would look something like: python liftOverPlink.py --map genotypes --out lifted --chain hg19ToHg38.over.chain.gz liftOverPlink.py [-h] -m MAPFILE [-p PEDFILE] [-d DATFILE] -o PREFIX -c CHAINFILE [-e LIFTOVEREXECUTABLE] wget hgdownload.soe.ucsc.edu/goldenPath/hg18/liftOver/hg18ToHg19.over.chain.gz hg18ToHg17.over.chain.gz 03-Apr-2006 10:17 231K hg18ToHg19.over.chain.gz 26-Jul-2010 11:25 137K hg18ToHg38.over.chain.gz 19-Feb-2014 16:21 336K |
|
|
|
Post by Tukuler al~Takruri on Jan 31, 2019 17:26:14 GMT -5
Amateurs have been making datasets for years. The idea is to pipe variously formatted data out into a chosen format. 'We' need to develop algorithms encode them build pgms bypassing systemic racialist givens under the code. Your reference source -- no matter how finely selected, quantity balanced, and scrubbed -- will make little diff if still running a 'Nicky P' pgm. 'We' have to build an algorithm to discern 'OoA' genome -- which also NeverLeftAfrica -- from Irani, Anatoli, or Sardine genomes that They say through out the continent and time depth of Hora Girl. Whether current African molecular biologists or some ESR lurker takes that career path in the future, academic credentialed heads will do it. Meanwhile, build us a base and keep pop numbers balanced when you run those pops actually relevant to the questions at hand. Keep on keeping on! Yeah! We have ADMIXTURE down to some extent but we need to create our own datasets. That means making sure all the popualtions in the dataset is align with the same hg19 or h18 or GrCh38 reference. Conversion back and forth must be done correctly conversion tools needed ---------- I may have to use PlinkLiftOver/LiftOver for my datasets prior to GrCh38 especially those obtained prior to 2013...like the Henn Qatari dataset? On Linux this would look something like: python liftOverPlink.py --map genotypes --out lifted --chain hg19ToHg38.over.chain.gz liftOverPlink.py [-h] -m MAPFILE [-p PEDFILE] [-d DATFILE] -o PREFIX -c CHAINFILE [-e LIFTOVEREXECUTABLE] wget hgdownload.soe.ucsc.edu/goldenPath/hg18/liftOver/hg18ToHg19.over.chain.gz hg18ToHg17.over.chain.gz 03-Apr-2006 10:17 231K hg18ToHg19.over.chain.gz 26-Jul-2010 11:25 137K hg18ToHg38.over.chain.gz 19-Feb-2014 16:21 336K |
|
|
|
Post by djoser-xyyman on Feb 1, 2019 10:24:01 GMT -5
I agree. I came across some tools that was NOT written or Nicky P had oversight to. I will post on it soon once I master some aspects. I agree his software may have bugs or code in it to skew results. I now realize also that even the hg19 thing is a sham. It means nothing as far as population affinity. Eg hg19 is made of selective sections of Europeans/African/Asian genes. With Europeans having the lions share. With Europeans being the youngest population. In other words ANYONE can make up reference(hgxx) and run with it. But it is needed for collaborative work. That is why an African reference can become very important...and why STRs are still important.
So we need to Look at other tools NOT written by Nicky P and that Max Planck group of racist. There are quite a few out there...but it is a lot of work getting on top of these new tools. Many are from 'independent" European Universities
|
|
|
|
Post by Tukuler al~Takruri on Feb 1, 2019 14:57:52 GMT -5
I don't mean The P only. Just used his name. It's all o dem. The operating prejudices are institutional/systemic. Why Amazon face recognition often takes dark skin for male skin? Bias built into the algorithms. Not intentional skewing, just the way it is. Instance: y ppl believe there's such a thing as olive skin. Show me a group of 'unadmixed' Euros -- NW W S E C -- with skin the color of any olive. Olives are like our Tricolor Flag: red black and green. As a people, we Afrikans have recognized ourselves as red and black since time immemorial. We are the factual olive skins.  I agree. I came across some tools that was NOT written or Nicky P had oversight to. I will post on it soon once I master some aspects. I agree his software may have bugs or code in it to skew results. I now realize also that even the hg19 thing is a sham. It means nothing as far as population affinity. Eg hg19 is made of selective sections of Europeans/African/Asian genes. With Europeans having the lions share. With Europeans being the youngest population. In other words ANYONE can make up reference(hgxx) and run with it. But it is needed for collaborative work. That is why an African reference can become very important...and why STRs are still important. So we need to Look at other tools NOT written by Nicky P and that Max Planck group of racist. There are quite a few out there...but it is a lot of work getting on top of these new tools. Many are from 'independent" European Universities |
|
|
|
Post by djoser-xyyman on Feb 19, 2019 8:45:50 GMT -5
As I said there are a few free software available to plot ADMIXTURE Charts. 1. PopHelper 2. R off the shelf 3. GenABEL 4. And this one I am using called Genesis. This seems to be the easiest and very user friendly The process: 1. Download/Obtain you dataset to compare 2. Convert these files to the ADMIXTURE format using PLINK software. PLINK is both Windows and Unix based. PLINK can convert VCF and other file formats to .bed, .bim and .fam formats which is the input format to ADMIXTURE. 3. The output formats from ADMIXTURE is .Q and .P . The .Q is the input format to Genesis. .P is used for PCA plotting. 4. Installation of Genesis is pretty straightforward since it is Windows based. Install JAVA JRE version 1.7 or 1.8. Make sure there is no JAVA duplicates or conflicts and make sure you JAVA PATH is correct(This can be the most difficult part). Double click on the Genesis.jar file and the Genesis program should start. Play with it. There are small example files included in the package to play with. 5. One time consuming bit is creating the “phenotype” files for GENESIS to run and sort. It is not needed but it is important to shift populations around. This file is absolutely needed to group and ungroup and sort files by Geographic regions or ethnic groups. The “phenotype” file can be manually created from your .fam file. But you need to use an editor like Notepad++ . It can also be done in Excel which is quicker but finalized in Notepad++ because Notepad++ can read and convert Unix to Windows. 6. Remember to use PLINK1.9 to merge, delete, add, and remove populations groups etc then convert to ADMIXTURE input files .bim, .fam, .bed. Remember ALL populations must first be aligned using the same reference so you are comparing apples-with-apples. Here is an example. www.bioinf.wits.ac.za/software/genesis/The main distribution includes documentation in PDF, HTML and info format. Amateur videos showing use of Genesis for PCA and admixture are also available. The instructional video structure.is-found.org/Genesis-admxiture.mov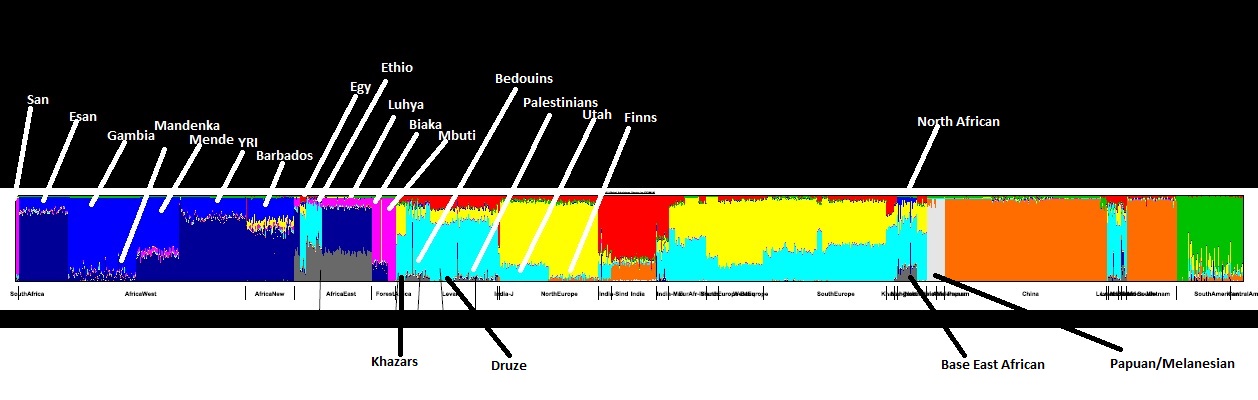 What does the image above show? Exactly what I have speculated for many years. 1. light blue is a base West African component NOT found East African. So much for Bantu Expansion. 2. YRI is distinct from Mende and Gambians 3. Finns are distinct from Utah Whites 4. North Africans carry base East African and SSA component 5. Modern Egyptians(these samples) are heavily admixed with Europeans but carry the base East African component 6. Bedouins and Palestinians carry Base East African component NOT found in Druze and Khazars 7. Mbuti and San are heavily related. Biaka and Mbuti are not closely related. San and Mbuti ar Read more: egyptsearchreloaded.proboards.com/thread/2861/got-playing-admixture?page=1#ixzz5fshQeqh9 |
|
|
|
Post by djoser-xyyman on Feb 19, 2019 11:14:10 GMT -5
plink --bfile jjk2888-jk-2134-jk-2911-out --recode --allow-no-sex --out Abusir-combined-PED
PLINK v1.90b6.7 64-bit (2 Dec 2018)
Options in effect:
--allow-no-sex
--bfile jjk2888-jk-2134-jk-2911-out
--out Abusir-combined-PED
--recode
Hostname: XXXX
Working directory: C:\XXXXX\plink
Start time: Tue Feb 19 11:04:49 2019
Random number seed: 1550592289
8083 MB RAM detected; reserving 4041 MB for main workspace.
90050 variants loaded from .bim file.
3 people (0 males, 0 females, 3 ambiguous) loaded from .fam.
Ambiguous sex IDs written to Abusir-combined-PED.nosex .
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 3 founders and 0 nonfounders present.
Calculating allele frequencies... done.
Warning: Nonmissing nonmale Y chromosome genotype(s) present; many commands
treat these as missing.
Total genotyping rate is 0.456989.
90050 variants and 3 people pass filters and QC.
Note: No phenotypes present.
--recode ped to Abusir-combined-PED.ped + Abusir-combined-PED.map ... done.
End time: Tue Feb 19 11:04:49 2019
|
|
|
|
Post by djoser-xyyman on Feb 19, 2019 12:51:21 GMT -5
Ok trying to merge 3 Abusir with the dataset I created above
Code
plink --bfile main-sample-admixture-test-set --bmerge jjk2888-jk-2134-jk-2911-out.bed jjk2888-jk-2134-jk-2911-out.bim jjk2888-jk-2134-jk-2911-out.fam --make-bed --out merge-abusir-global
PLINK v1.90b6.7 64-bit (2 Dec 2018)
Options in effect:
--bfile main-sample-admixture-test-set
--bmerge jjk2888-jk-2134-jk-2911-out.bed jjk2888-jk-2134-jk-2911-out.bim jjk2888-jk-2134-jk-2911-out.fam
--make-bed
--out merge-abusir-global
Hostname: XXXX
Working directory: C:\XXXX\plink
Start time: Tue Feb 19 12:36:52 2019
Random number seed: 1550597813
8083 MB RAM detected; reserving 4041 MB for main workspace.
2447 people loaded from main-sample-admixture-test-set.fam.
3 people to be merged from jjk2888-jk-2134-jk-2911-out.fam.
Of these, 3 are new, while 0 are present in the base dataset.
Warning: Multiple positions seen for variant '.'.
Warning: Multiple positions seen for variant '.'.
warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
135056 markers loaded from main-sample-admixture-test-set.bim.
90050 markers to be merged from jjk2888-jk-2134-jk-2911-out.bim.
Of these, 1 is new, while 90049 are present in the base dataset.
Error: 1 variant with 3+ alleles present.
* If you believe this is due to strand inconsistency, try --flip with merge-abusir-global-merge.missnp.
(Warning: if this seems to work, strand errors involving SNPs with A/T or C/G alleles probably remain in your data. If LD between nearby SNPs is high,
--flip-scan should detect them.)
* If you are dealing with genuine multiallelic variants, we recommend exporting that subset of the data to VCF (via e.g. '--recode vcf'), merging with
another tool/script, and then importing the result; PLINK is not yet suited to handling them.
End time: Tue Feb 19 12:36:58 2019
|
|
|
|
Post by djoser-xyyman on Feb 19, 2019 14:09:37 GMT -5
FYI
PLINK(1.9) PLINK2.0, Samtools can be run from Windows cmd.exe
Download windows version and run from DOS CMD terminal. Make sure you are in correct folder containing the samtools and/or PLINK.
In other words most of you dataset creation/manipulation can be down in MicroSoft Windows/CMD but the ADMIXTURE must be run in UNIX/Linux/Ubuntu. There is no Windows based version of ADMIXTURE I am aware of.
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 10:09:24 GMT -5
I believe this is the final step.
Looks like one variant needs to be removed then I can merge with my base dataset I created.
ERROR QUOTE:
----------------------------------------------------------------------------------
warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
135056 markers loaded from main-sample-admixture-test-set.bim.
90050 markers to be merged from jjk2888-jk-2134-jk-2911-out.bim.
Of these, 1 is new, while 90049 are present in the base dataset.
Error: 1 variant with 3+ alleles present.
-----------------------------------
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 10:11:00 GMT -5
I need to figure out how to remove that one problem variant/SNP.
Looking at some commands and scripts now.......
Suggestions
Try:
--biallelic-only strict
Or
Not sure about your exact question, but in VCFtools you can quickly filter for only bi-allelic sites using:
vcftools --vcf_file1.vcf --min-alleles 2 --max-alleles 2 --recode --out output_file_name.vcf
|
|



