|
|
Post by djoser-xyyman on Feb 20, 2019 16:55:38 GMT -5
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 18:28:20 GMT -5
folder~/working$ vcftools --vcf jk2888-jk-2134-jk-2911-out.vcf --min-alleles 2 --max-alleles 2 --recode --out jk2888-jk-2134-jk-2911-out-fix.vcf
VCFtools - 0.1.15
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--vcf jk2888-jk-2134-jk-2911-out.vcf
--max-alleles 2
--min-alleles 2
--out jk2888-jk-2134-jk-2911-out-fix.vcf
--recode
After filtering, kept 3 out of 3 Individuals
Outputting VCF file...
After filtering, kept 89975 out of a possible 90050 Sites
Run Time = 14.00 seconds
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 20:27:19 GMT -5
vcftools --gzvcf jk2888-jk-2134-jk-2911-out.vcf.gz --min-alleles 2 --max-alleles 2 --recode --out jk2888-jk-2134-jk-2911-out-fix3.vcf
VCFtools - 0.1.15
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--gzvcf jk2888-jk-2134-jk-2911-out.vcf.gz
--max-alleles 2
--min-alleles 2
--out jk2888-jk-2134-jk-2911-out-fix3.vcf
--recode
Using zlib version: 1.2.11
After filtering, kept 3 out of 3 Individuals
Outputting VCF file...
After filtering, kept 89975 out of a possible 90050 Sites
Run Time = 15.00 seconds
----------
vcftools --vcf jk2888-jk-2134-jk-2911-out.vcf --min-alleles 2 --max-alleles 2 --recode --out jk2888-jk-2134-jk-2911-out-fix2.vcf
VCFtools - 0.1.15
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--vcf jk2888-jk-2134-jk-2911-out.vcf
--max-alleles 2
--min-alleles 2
--out jk2888-jk-2134-jk-2911-out-fix2.vcf
--recode
After filtering, kept 3 out of 3 Individuals
Outputting VCF file...
After filtering, kept 89975 out of a possible 90050 Sites
Run Time = 14.00 seconds
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 22:10:29 GMT -5
Finally got it!!!!!!Use plink vcf to get the abusir bim, fam and bed files. First remove trialleles - vcftools --gzvcf jk2888-jk-2134-jk-2911-out.vcf.gz --min-alleles 2 --max-alleles 2 --recode This removed the errors like multi allielic and bad variants. Run through ADMIXTURE to get the Q file to plot, I will then merge with many other datasets 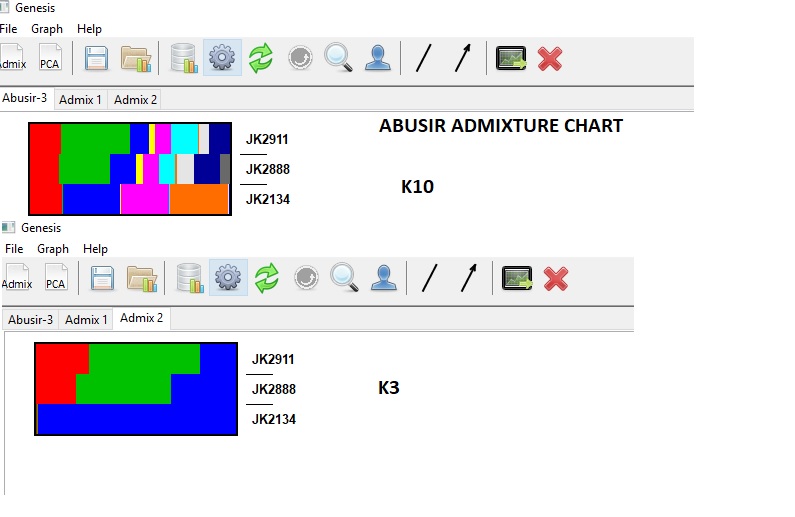
|
|
|
|
Post by djoser-xyyman on Feb 20, 2019 22:12:43 GMT -5
Apparently - JK2134 is different to the other Abusir. I will now merge with my dataset above.
|
|
|
|
Post by djoser-xyyman on Feb 21, 2019 8:27:30 GMT -5
From combined VCF Abusir file after they were aligned with the hg19 using FreeBayes to align 1. Remove problem variants/SNPs using VCFtools vcf --min-alleles 2 --max-alleles 2 –recode. The output is a clean Abusir VCF file that can be processed in PLINK1.9 or PLINK2 2. With the ‘clean’ Abusir VCF file use PLINK to create your input files for ADMIXTURE ie .bim, .bed, .fam. You can use Notepad++ to create a phenotype(.phe) file from the .fam file you create. A Phenotype file is needed if you want to sort/group individuals or population is specific order or your taste. Copy your .fam file to a .phe file then open in notepad++ and names or continents to each individual. 3. Now use ADMIXTURE to run you Abusir .bim, .bed, .fam file from K2 to K10 Create .Q and .P files. 4. Use Genesis, PopHelper or GenABEL, R etc to plot your ADMIXTURE .Q files. Genesis is great because it is windows/java driven so you can right and left click to move population groups around. Delete populations also if you need to.  |
|
|
|
Post by djoser-xyyman on Feb 22, 2019 8:49:04 GMT -5
To those who can follow. Those color codes don’t mean anything until the Abusirs are combined/merged with the Global Populations The K values obtained through ADMIXTURE. But as you can see the one of the Abusir is significantly different to the other two. This has nothing to do with “Eurasian” vs “African”. It could mean one was a Berber and the other two Sudanese related. Or One Sudanese and the other two Somalian related…..chose your population. I am working through merging There are problem SNPs I am trying to work through. Apparently the Abusir has much LESS SNPs when aligned/merging to the Global populations.
It can also mean population change over between the two and the one. Therefore the time period(chronology) has to be taken into account.
|
|
|
|
Post by djoser-xyyman on Feb 25, 2019 10:59:11 GMT -5
placeholder ----------------------- quote " bcftools norm --multiallelics '-any' a.vcf | bcftools norm -f '/path/to/genome.fa' > a.normed.vcf | | After I did it for both files, I merge them using bcftools merge. " I'm not sure about the following: if you use bcftools merge to merge vcf.gz files does it merge the overlapping variants only or does it also merge variants that are present in one dataset but not in the other? I'm not aware of such in option. If you can make sure, that in your vcf file you like to merge, are no genotypes ./., you could filter out those sites after merging. bcftools merge in1.vcf.gz in2.vcf.gz|bcftools filter -e 'GT="./."' > out.vcfIf you are interested to find out which variants are in all your vcf files, without merging them than the term you are looking for is intersect.# Extract and write records from A shared by both A and B using exact allele match bcftools isec A.vcf.gz B.vcf.gz -p dir -n =2 -w 1samtools.github.io/bcftools/bcftools.html#normCommands used to align and normalize ‘different” datasets are: 1. bcftools norm -f ref.fa in.vcf -O z > out.vcf.gz 2. java -jar GenomeAnalysisTK.jar -T LeftAlignAndTrimVariants --trimAlleles -R ref.fa --variant in.vcf.gz -o out.vcf.gz 3. vt normalize -r ref.vcf.gz -o out.vcf.gz or 4. bcftools norm --multiallelics '-any' a.vcf | bcftools norm -f '/path/to/genome.fa' > a.normed.vcf |
|
|
|
Post by djoser-xyyman on Feb 27, 2019 9:08:16 GMT -5
Quote:
“Why don't you figure out the common snps between the two datasets using a shell command (awk) or a R one-liner. You can then reduce each of those datasets to the same set of SNPS using the "--extract" option and then merge the datasets. Also, you should check if the two datasets have the same build.”
quote:
"Hi I suggest to combine R & Plink this way:
> R code:
> map2 = read.delim("file2.map", header=F, quote="")
> map1 = read.delim("file1.map", header=F, quote="")
> common.snps = which(map2$V2 %in% map1$V2)
> write.table(map2$V2[common.snps], file="list.snps", sep="\t", col.names=F, row.names=F, quote=F )
and finally
> the Plink commands:
> plink --bfile <file1> --extract list.snps --make-bed --out data1
> plink --bfile <file2> --extract list.snps --make-bed --out data2
> plink --bfile data1 --bmerge data2.bed data2.bim data2.fam --make-bed --out merge"
|
|
|
|
Post by djoser-xyyman on Feb 27, 2019 9:38:17 GMT -5
PLINK
Pairwise IBD estimation
In a homogeneous sample, it is possible to calculate genome-wide IBD given IBS information, as long as a large number of SNPs are available (probably 1000 independent SNPs at a bare minimum; ideally 100K or more).
code
plink --file mydata –genome
which creates the file <plink.genome>
**Segmental sharing: detection of extended haplotypes shared IBD**
There are five important steps to this analysis:
Obtain a homogeneous sample
Remove very closely related individuals
Prune SNP set
Detect segments
Associate with disease
generate a plink.genome file. This will be used subsequently in a number of steps.
--cluster (cluster individuals)
--matrix (generate .mdist file, used to generate MDS plots)
--ppc (threshold for PPC test, not to cluster individuals)
--mds-plot (generate a multidimensional scaling plot)
--ibs-test (as case/control less similar on average?)
--neighbour (option to find individual outliers)
Detecting shared segments (extended, shared haplotypes)
The segmental sharing analysis requires approximately independent SNPs (i.e. linkage equilibrium).
With a newly pruned fileset, ideally containing only independent, high quality SNPs in individuals who are not very closely related but are from the same population, run the command
plink --bfile mydata4 --read-genome plink.genome –segment
PLINK expects the 3rd column the MAP/BIM file to contain genetic distances in Morgan units.
The --segment option generates a file
“plink.segment”
|
|
|
|
Post by djoser-xyyman on Mar 1, 2019 11:44:15 GMT -5
Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods Verena J. Schuenemann Generation of nuclear data quote: "In order to analyse the nuclear DNA we selected 40 samples with high mtDNA coverage and low mtDNA contamination. Using in solution enrichment for 1.2 million genome-wide SNPs26, we obtained between 3,632 and 508,360 target SNPs per sample Overall, the nuclear DNA showed poor preservation compared to the mtDNA as depicted by a high mitochondrial/nuclear DNA ratio of on average around 18,000. In many samples, nuclear DNA damage was relatively low, indicating modern contamination. Three out of 40 samples fulfilling these criteria had acceptable nuclear contamination rates Two samples from the Pre-Ptolemaic Periods (New Kingdom to Late Period) had 5.3 and 0.5% nuclear contamination and yielded 132,084 and 508,360 SNPs, respectively, and one sample from the Ptolemaic Period had 7.3% contamination and yielded 201,967 SNPs. As shown below, to rule out any impact of potential contamination on our results, we analysed the three samples separately or **replicated results using only the least contaminated** sample.We extracted DNA from 151 mummified human remains and prepared double-stranded Illumina libraries with dual barcodes Nuclear DNA capture The non-UDG and UDG treated libraries were enriched by hybridization to probes targeting approximately 1.24 million genomic SNPs as described previously25. The target SNPs consist of panels 1 and 2 as described in Mathieson et al.41 and Fu et al.26 (see Supplementary Note 2 for details). Three samples were selected for down-stream analysis: JK2134, JK2888 and JK2911. In all three of these samples, contamination estimates were acceptable Nuclear data analysis: genotyping We called genotypes from the UDG treated data for the three individuals by sampling a random read per SNP in the SNP-capture panel, using a custom tool ‘pileupCaller’, available at Data availabilityThe mapped BAM files for the 90 mitochondrial samples and three nuclear samples are deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena) with the study ID ERP017224. www.ebi.ac.uk/ena/data/search?query=ERP017224" " |
|
|
|
Post by djoser-xyyman on Mar 4, 2019 9:47:39 GMT -5
gatk --java-options "-Xmx8G" HaplotypeCaller -R reference.fasta -I input.bam -O output.vcf
gatk HaplotypeCaller -R hg19.fa -I JK2134.bam -O JK2134-bam.vcf
samtools index ~/workingfolder/JK2888.bam
samtools index ~/workingfolder/JK2911.bam
gatk HaplotypeCaller -R hg19.fa -I JK2911.bam -O JK2911-bam.vcf
gatk HaplotypeCaller -R hg19.fa -I JK2888.bam -O JK2888-bam.vcf
bgzip JK2134-bam.vcf
bgzip JK2888-bam.vcf
bgzip JK2911-bam.vcf
tabix -p vcf JK2911-bam.vcf.gz
tabix -p vcf JK2134-bam.vcf.gz
tabix -p vcf JK2888-bam.vcf.gz
MERGING Abusir WITH my Dataset
vcf-merge abusir-bam.vcf.gz main-sample-admixture-test-set.vcf.gz | bgzip -c > abusir-global.vcf.gz
detected that all genotypes are missing for a SNP locus.
Please apply quality-control filters to remove such loci.
plink --bfile abusir-global --geno 0.999 --make-bed --out abusir-global-fix
admixture abusir-global-fix.bed 2plink --vcf abusir-global.vcf.gz --const-fid 0 --make-bed --out abusir-global
CONVERT VCF to bed in PLINK
jk2888-jk-2134-jk-2911-out.vcf
plink --vcf jk2888-jk-2134-jk-2911-out.vcf --make-bed --out abusirpop
|
|
|
|
Post by djoser-xyyman on Mar 4, 2019 10:41:55 GMT -5
Finally. My trek is over. So one Abusir jk2134 is non-African(contamination reading the preamble?) and the other two are pure Africans(red). More African than even Afro-barbados and Ethiopians. This is impossible. All 3 has to be African not just two!!! Creating the Admixture charts is outlined above(coding). It is simpler than I thought. 1. Download bam files of the Abusir 2. Index with samtools 3. Align to hg19(abusir) 4. Create your global compare datasets. Also align with hg19. 5. Merge Abusirs with the "YOUR" globals datasets. 6. Create your abusir-global dataset and create plink bed files 7. run the plink bed files through admixture 8. plot your admixture files with genesis or some such tool. This is my K2 I will clean up and plot K3-K10 with clean labels. Important point to note is using different source global datasets and overlapping SNPs give different results. 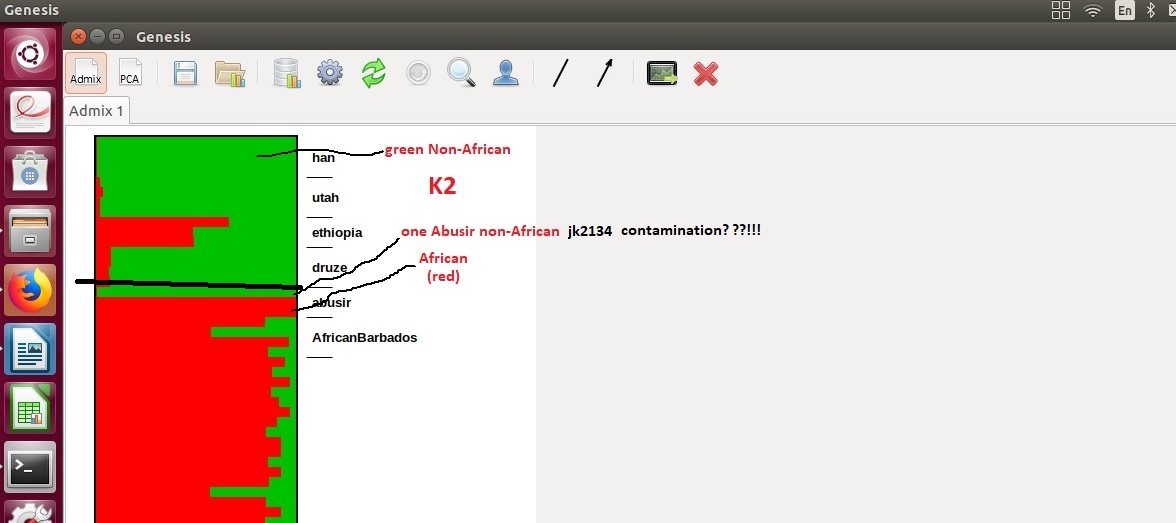 |
|
|
|
Post by djoser-xyyman on Mar 9, 2019 14:01:33 GMT -5
@ K5. Abusir are far from being related to West Asian. They are old Africans I will explain. . I will provide more plots but this is a sample. JK2134 is an older African bloodline  |
|
|
|
Post by djoser-xyyman on Mar 9, 2019 22:27:38 GMT -5
You asked a good question in a prior post.(oh there was never a movement of humans back into Africa until the Ottoman Turks. There was no movement of variants back into Africa either. That type of gabble-gook comes from ElMeastro) Anyways. To your question. Why is my analysis "better" than Schuenemann? Neither is better. Two different approaches. Schuenemann's is designed for deception. quote from Schuenemann..."The non-UDG and UDG treated libraries were enriched by hybridization to probes targeting approximately 1.24 million genomic SNPs as described previously25. The target SNPs consist of panels 1 and 2 as described in Mathieson et al.41 and Fu et al.26 (see Supplementary Note 2 for details)." What does it mean and what did she do? Schuenemann targeted specific SNPs in the human genome (panels 1 and 2). They were NOT randomly chosen or across the entire human genome. Why? This not rocket science. What I did was took what they gave us(variants/SNP) and compare it with a different global dataset ie population group. I included other populations besides those from HGDP/ I got some from Henn, University of Barcelona and Doron Behar Jews dataset. They showed a totally different outline than Scheuermann's. She stacked the deck to skew the results AWAY from SSA. Abusir are undoubtedly African. ALL OF THEM. Including JK2134. In fact JK2134 carry OLDER VAriants/SNP than the other two that is why it appears so different to the other two. I was surprised to find so much American ancestry in jk2134 but absent in the other two. That pink(and yellow) ancestry is NOT found in Bedouins!!!! Surprisingly yellow and pink is not found in Bedouins but found in Makrani. In fact Makrani carries even the "European" green along with pink and yellow and SSA blue. Makrani carry majority European green. This is most likely the Neolithic package. That left Africa and entered Eurasia what is a variant?? The human genome carry about 3 BILLION base pairs. Of those 3 BILLION base pairs about 10 Million are different. ie variants or SNPs. In short of 3,000,000,000 base pairs 3,000,000,000 -10,000,000 =2,990,000,000 bases are the SAME in all humans. We are more similar than different!!!! News flash!! We differ through variants or SNPs. But of the 10,000,000 SNPs only are few are routinely tested in genomic analysis. I chose a larger/different/more random/more transect set of SNPs than Schuenemann. She skewed the results. Plus she limited SSA to one group of West Africans...the dreaded YRI.
Understand how ADMIXTURE works. The intent is to include SNPs that "overlap". So the more populations the more accurate. The more SNPs also the more accurate. She did neither.
I think back on ES you said that "back migration" was simply the movement of African variants back into Africa. If so can you expand on this a bit more, such as what sets the border of a genetic variant?
|
|



