|
|
Post by djoser-xyyman on Mar 10, 2019 6:18:06 GMT -5
This is huge so a cut it down to size. I have to figure out how to upload the entire file. I manually cut this down to size and remove some similar populations,  |
|
|
|
Post by djoser-xyyman on Mar 11, 2019 12:30:54 GMT -5
So what is this telling us?
1. Over 1 million SNPs/Variants were recovered from the ancient Africans
2. Only 135057 SNPs/Variants (over X10) recovered from the Abusir average overlap
3. Since we can only merge variants from “overlapping” datasets that means only 135057 variants will be merged from the Abusir/global/ancient Africans. The Abusir are the rate determining step …ie the bottleneck ie the limitation. The fewest varaiants are from the 3Abusir.
So it seems it is possible for me to merge and perform ADMIXTURE on the 3 Abusir, 16 Ancient Africans and my global dataset of about 2400 modern humans from across the globe
More to come….
--------------------------------------------
plink --bfile skoglund16africans --bmerge abusir-global-fix.bed abusir-global-fix.bim abusir-global-fix.fam --make-bed --out abusir-global-Skog
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
1233013 markers loaded from skoglund16africans.bim.
135162 markers to be merged from abusir-global-fix.bim.
Of these, 3823 are new, while 131339 are present in the base dataset.
Error: 24407 variants with 3+ alleles present.
-------------------------------------------------
plink --bfile abusir-global-fix --bmerge skoglund16africans.bed skoglund16africans.bim skoglund16africans.fam --make-bed --out abusir-global-Skog2
Working directory: C:\XXXXXX\plink
Start time: Mon Mar 11 13:13:23 2019
Random number seed: 1552324403
8083 MB RAM detected; reserving 4041 MB for main workspace.
2450 people loaded from abusir-global-fix.fam.
16 people to be merged from skoglund16africans.fam.
Of these, 16 are new, while 0 are present in the base dataset.
Warning: Multiple positions seen for variant '.'.
Warning: Multiple positions seen for variant '.'.
Warning: Multiple positions seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
Warning: Multiple chromosomes seen for variant '.'.
135057 markers loaded from abusir-global-fix.bim.
1233013 markers to be merged from skoglund16africans.bim.
Of these, 1101779 are new, while 131234 are present in the base dataset.
Error: 24407 variants with 3+ alleles present.
* If you believe this is due to strand inconsistency, try --flip with
abusir-global-Skog2-merge.missnp.
(Warning: if this seems to work, strand errors involving SNPs with A/T or C/G
alleles probably remain in your data. If LD between nearby SNPs is high,
--flip-scan should detect them.)
* If you are dealing with genuine multiallelic variants, we recommend exporting
that subset of the data to VCF (via e.g. '--recode vcf'), merging with
another tool/script, and then importing the result; PLINK is not yet suited
to handling them.
End time: Mon Mar 11 13:13:26 2019
|
|
|
|
Post by zarahan on Mar 12, 2019 9:18:01 GMT -5
Understand how ADMIXTURE works. The intent is to include SNPs that "overlap". So the more populations the more accurate. The more SNPs also the more accurate. She did neither.LOL why are we not surprised.. THey have been running the rigged sampling game for decades.. But we have to keep relentlessly pointing it out so that distorted parts of their narrative are exposed and debunked. There is a new generation of young blacks out there, some brainwashed children of the media, and some of whom, alas are not interested in serious reading, analysis and reflection on these things. But for those interested, they will have to carry the battle forward, and they will see the same distortion tactics. Again for the noobs.. you will see different versions, but the same tactics 30 years hence. Remember you saw it here first.. lol QUICK SNAPSHOT: Stacked decks and rigged samples in studies of Africa and the Nile Valley... 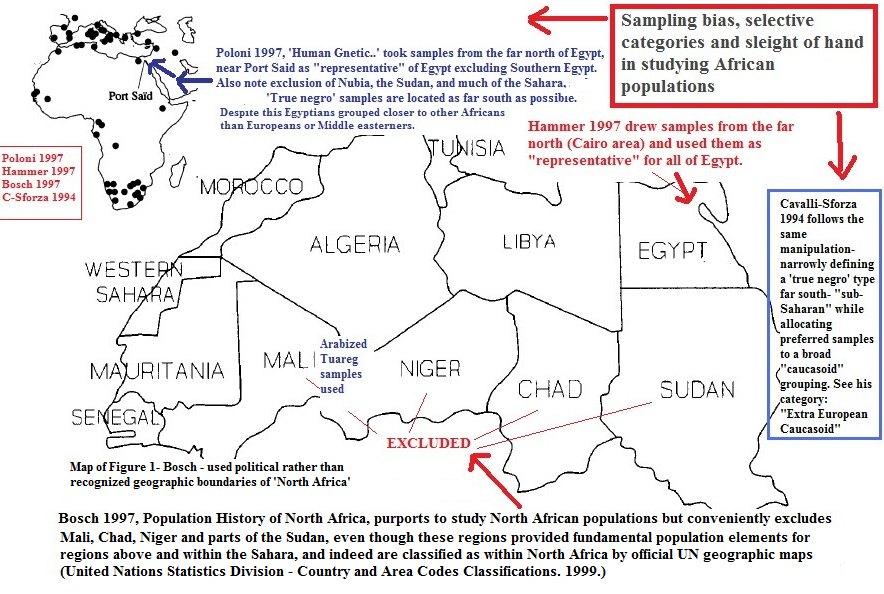  ---------------------------------------------------------------------------------------------------  ------------------------------------------------------------------------- So it seems it is possible for me to merge and perform ADMIXTURE on the 3 Abusir, 16 Ancient Africans and my global dataset of about 2400 modern humans from across the globe
More to come…. Keep up the good work. It will be interesting to see some of these links into the Nile Valley 20 years on.. Reloaded will have no doubt disappeared by then. Still, looking at the Biblical picture which Diop considered to be a valid line of background cultural evidence: SONS OF HAM -- Mizraim- Egypt -- Cush- usually refers to Sudanic zone near Egypt and less used -Mesopotamia/Ethiopia/Yemen and perhaps extending down to the Horn -- Phut- Somalia, Southern Sudan or Eritrea Libya? -- Caanan- the Palestine, Lebanon area Sons of Mizraim (Egypt): Ludim, Anamim, Lehabim, Naphtuhim, Pathrusim, Casluhim (out of whom came Philistim), Caphtorim. We already know how Haplogroup E, originating in Africa, on the Y-DNA side unites 70% of all males on the African continent, with some overlap into other adjacent areas, including Arabia. 20 or 50 years hence it will be interesting to see if Moses' framework of peoples and relationships above is confirmed. We can't trace all specific ancient groups of course, but we can get a better idea of linkages and relationships among peoples of the broad "Near East" region in/near Egypt, the Horn and even the closer-by Mediterranean zone. So far what Moses generally wrote has held true, the close relationships between Cush and Mizraim (brothers) for example, and the linkages between all these peoples, versus some "splittist" models of Eurocentrism. The "Ludim" are mentioned in Jeremiah as those famed who "handle the bow." I wonder if said Ludim might relate to groups farther south, some Nubians who in olden time were famed as archers. The anthro would confirm close relationships between Nubia and Kemet of course.
|
|
|
|
Post by djoser-xyyman on Apr 2, 2019 19:07:02 GMT -5
This is the problem! How can SSA DNA exist in Armenia 3800bc but not present IN Africa(North) 300ad???!!! Makes Absolutely no sense. African DNA in Eurasia but NOT in North Africa. THEY ARE LYING ...as usual. or twisting the data. Genetic evidence for an origin of the Armenians from Bronze Age mixing of multiple populations -Marc Haber, 2016 Quote: "Haber et al(2016) "We sought to date these mixture of events using exponential decay of admixture-induced LD. The oldest mixture events appear to be between populations related to sub-Saharan Africans and West Europeans occurring ~3800 BCE, followed closely by a mixture of Sardinian and Caucasus-related populations. Later, several mixture events occurred from 3000 to 1200 BCE involving diverse Eurasian populations (Table 1, Figure 3)." "  Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods - Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-Roman periods -
Verena J. Schuenemann,quote: "However, we note that all our genetic data were obtained from a single site in Middle Egypt and may not be representative for all of ancient Egypt. It is possible that populations in the south of Egypt were more closely related to those of Nubia and had a higher sub-Saharan genetic component, in which case the argument for an influx of sub-Saharan ancestries after the Roman Period might only be partially valid and have to be nuanced. Clearly, more genetic studies on ancient human remains from southern Egypt and Sudan are needed before apodictic statements can be madeIn particular, we seek to determine if the inhabitants of this settlement were affected at the genetic level by foreign conquest and domination, especially during the Ptolemaic (332–30BCE) and Roman (30BCE–395CE) Periods." THESE EUROPEANS CAN'T HELP THEMSELVES WITH THEIR LIES AND DISTORTIONS |
|
|
|
Post by Tukuler al~Takruri on Apr 2, 2019 19:46:04 GMT -5
Hahahahahaa!
U still payin tension 2 they interpretations?
XD 😝 XD
|
|
|
|
Post by samuel on Apr 23, 2019 16:54:08 GMT -5
Really makes you wonder why they are pulling the wool over our eyes? What do they have to fear when it becomes known the real race of the AE? Self esteem for black people. Knowing they weren't always thugs and rappers and slaves. Why do they fear this? This is the LIBERAL ESTABLISHMENT mind you.
|
|
|
|
Post by kel on Apr 24, 2019 6:48:16 GMT -5
Um......ok (eyeroll)
|
|
|
|
Post by zarahan on May 4, 2019 23:11:33 GMT -5
xyz quoted from Schuemann: quote:
"However, we note that all our genetic data were obtained from a single site in Middle Egypt and may not be representative for all of ancient Egypt. It is possible that populations in the south of Egypt were more closely related to those of Nubia and had a higher sub-Saharan genetic component, in which case the argument for an influx of sub-Saharan ancestries after the Roman Period might only be partially valid and have to be nuanced.
Clearly, more genetic studies on ancient human remains from southern Egypt and Sudan are needed before apodictic statements can be made LOL, at their shallow "spin" attempt. NO we don;t need more genetic studies before "apodictic" (self evident absolute) statements can be made. We already have plenty of evidence- dental, cranial, limb proportion, archaeo/cultural, etc. We don;t need to wait around for ancient DNA evidence. Credible research for decades already tells us a lot about ancient Upper Egypt and its population. We don't need to wait around for another obvious "spin" attempt.. lol  |
|
|
|
Post by zarahan on May 4, 2019 23:12:56 GMT -5
This is the problem! How can SSA DNA exist in Armenia 3800bc but not present IN Africa(North) 300ad???!!! Makes Absolutely no sense. African DNA in Eurasia but NOT in North Africa. THEY ARE LYING ...as usual. or twisting the data. Genetic evidence for an origin of the Armenians from Bronze Age mixing of multiple populations -Marc Haber, 2016 Quote: "Haber et al(2016) "We sought to date these mixture of events using exponential decay of admixture-induced LD. The oldest mixture events appear to be between populations related to sub-Saharan Africans and West Europeans occurring ~3800 BCE, followed closely by a mixture of Sardinian and Caucasus-related populations. Later, several mixture events occurred from 3000 to 1200 BCE involving diverse Eurasian populations (Table 1, Figure 3)." Hmm, this might explain why a 1990 Encyclopedia Britannica 'Macropedia' article I remember on Egypt dubbed some ancient remains as resembling "Armenoids." I did a double take at what the mysterious term "Armenoids" meant, but it just might be that the remains were partly sub-Saharan resemblance but they had to put another label on it. Its like a 1970s article on Ethiopia describing the inhabitants as " a white people with black skin!" If they were "white", how could they have black skin? But where did these part negro mix "Armenoids" come from? Its not an OOA or Natufian era thing. The dates given are relatively recent- 3800BCE.. |
|
|
|
Post by djoser-xyyman on May 28, 2019 5:53:05 GMT -5
Place holder --------------------- Population genetics data for 22 autosomal STR loci in European, South Asian and African populations using SureID® 23comp Human DNA Identification Kit Author links open overlay panelSasitaranIyavooaOlatundeAfolabiaByronBoggiaAusmaBernotaitebThomasHaizela Show more doi.org/10.1016/j.forsciint.2019.05.033Get rights and content Abstract Allele frequency data for 22 short tandem repeat loci; D18S1364, D1S1656, D13S325, D5S2800, D9S1122, D4S2366, D3S1744, D12S391, D11S2368, D21S2055, D20S482, D8S1132, D7S3048, D2S441, D19S253, D10S1248, D17S1301, D22-GATA198B05, D16S539, D6S474, D14S1434 and D15S659 from the SureID® 23comp Human DNA Identification Kit have been determined for unrelated individuals in European, South Asian and African populations. Deviations from Hardy–Weinberg equilibrium were observed in loci D1S1656 and D19S253 in European; D18S1364, D6S474 and D14S1434 in South Asian; and D9S1122 and D8S1132 in African populations (p-value < 0.05). However, after Bonferroni correction no significant deviations were observed (p-value < 0.002). The most discriminating loci were D1S1656 and D12S391 for European (PD = 0.977), D21S2055 for South Asian (PD = 0.980), and D21S2055 and D7S3048 for African (PD = 0.972) populations. The match probabilities were 1 in 6.7 × 1025 for European, 1 in 1.4 × 1026 for South Asian and 1 in 1.6 × 1026 for African populations. These findings established the high discriminatory capacity and robustness of the tested STR loci for forensic identification and kinship testing. Keywords Short tandem repeatsSureID® 23comppopulation genetics dataEuropeanSouth AsianAfricanallele frequencies |
|
|
|
Post by djoser-xyyman on Dec 21, 2019 6:30:11 GMT -5
Another tool to look at. One of these days I will get the time to do a deep dive.
==============July 30, 2019Software Open Access
Genomic GPS: using genetic distance from individuals to public data for genomic analysis without disclosing personal genomes
Kunhee Kim; Hyungryul Baik; Chloe Soohyun Jang; Jin Kyung Roh; Eleazer Eskin; Buhm Han
Genomic GPS applies the multilateration technique commonly used in the global positioning system (GPS) to genomic data. In the framework we present here, investigators calculate genetic distances between their samples and reference samples, which are from data held in the public domain, and share this information with others. This sharing enables certain types of genomic analysis, such as identifying sample overlaps and close relatives, decomposing ancestry, and mapping of geographical origin without disclosing personal genome. Thus, our method can be seen as a balance between open data sharing and privacy protection.
|
|
|
|
Post by djoser-xyyman on Jan 18, 2020 11:12:53 GMT -5
Here is an idea. Write to the authors requesting the CODIS STRs. Remember they removed the STRs from the Abusirs before they made the genome freely available. By hiding that information is an indication they are being deceptive.
Remember JAMA listed the CODIS STRs of the Amarnas. Why didn't they do the same for the Abusir. Why, why did remove it? Obviously they don't want us to know.
You people. Flood their inbox and ask for the STR results for the Abusir.
|
|
|
|
Post by djoser-xyyman on Jan 18, 2020 11:16:54 GMT -5
And here is the kicker. Not 1 STR was removed. ALL were removed. ALL!!!!. Tsk! Tsk!
|
|
|
|
Post by djoser-xyyman on Feb 2, 2020 6:41:16 GMT -5
Placeholder
FrogAncestryCalc: A standalone batch likelihood computation tool for ancestry inference panels catalogued in FROG-kb
Haseena Rajeevan
Usha Soundararajan
Andrew J. Pakstis
Kenneth K. Kidd
Open AccessPublished:January 21, 2020DOI:https://doi.org/10.1016/j.fsigen.2020.102237
Highlights
•
FrogAncestryCalc is the stand-alone version of FROG-kb.
•
Simultaneously computes population likelihoods for multiple individuals.
•
Five Ancestry Inference (AI) panels are implemented.
•
Available for download from FROG-kb and GitHub.
•
FrogAncestryCalc is open source.
|
|
|
|
Post by djoser-xyyman on Feb 8, 2020 19:12:57 GMT -5
Here is another tool that may work.
Mining Whole Genome Sequence data to efficiently
attribute individuals to source populations
Francisco J. Perez-Reche ´
1,*, Ovidiu Rotariu2
, Bruno S. Lopes3
, Ken J. Forbes3
, and
Norval J.C. Strachan2
Computational time. The computational time for MMD is much shorter than STRUCTURE. Fig. 4(c) shows a comparison
of runtimes for self-attribution of Campylobacter isolates as a function of the number of SNP loci describing the genotypes.
The MMD is between 100 and 105
times faster than STRUCTURE for every run from 1 to ∼ 2×104 SNPs. Since the running
time of MMD increases slowly with the number of loci compared to that of STRUCTURE, the efficiency of MMD improves
relative to that of STRUCTURE for extended genotypes. For instance, STRUCTURE takes ∼ 40h to assign a 25 937 cgSNP
genotype whereas MMD completes the task in ∼ 0.57 seconds (MMD implementation in R67, Processor: Intel® CoreTM
i7-3770 3.40GHz).
The MMD method is around twice as fast as ADMIXTURE when considering the 659 276 SNP dataset. More explicitly,
MMD takes ∼ 15 seconds to assign an individual whereas ADMIXTURE takes ∼ 38 seconds to infer the ancestry of one
individual (times based on an Intel® CoreTM i7-3770 3.40GHz processor for both algorithms).
Results
Source attribution with the MMD method
We propose the Minimal Multilocus Distance (MMD) method to estimate the probability pu,s
that an individual u is attributed
to a population source s based on the similarity between the genotype of the individual to be attributed and genotypes from the
sources. The similarity between pairs of genotypes is quantified by the Hamming distance which simply gives the number of
loci at which the genotypes differ64. The smaller the distance between genotypes, the larger the probability that they originate
from the same source (see Methods). To test the accuracy of the MMD method, we studied self-attribution, a cross-validation
method65, 66 which consists in removing individuals from the source population and estimating the probability that they are
correctly attributed to their source based on the remainder5, 12, 13, 51 (Fig. 2).
|
|







