|
|
Post by djoser-xyyman on Dec 13, 2018 15:33:34 GMT -5
|
|
|
|
Post by djoser-xyyman on Jan 9, 2019 15:17:18 GMT -5
SNPedia Report on Abusir jk2888 Oh! CODIS STR feasture was REMOVED in this version. Can't get my hands on the prior version with CODIS STR feature. A veteran coder can reinstate that feature. I am a novice coder! BAM Analysis kit has pulled out these SNPs for Abusir-JK2888 Bam Kit is windows based and can be easily installed. 1. Download and install Bam Analysis kit(windows based but runs in DOS mode) www.y-str.org/2014/04/bam-analysis-kit.html2. Download Bam and Fastq files of the 3 Abusir mummies eg JK2888 etc 3. Make sure all files are in the same folder!!!! Including data files 4. Crate a sub-folder named "out". THIS IS VERY IMPORTANT! All Lower case "out" 5. Run the first Abusir bam file. Eg jk-2888. Accept all default settings if you want 6. The program will run for about 1-2hrs creating VCF files for each chromosome which you can copy and save as they are created. After about 2 hrs the "out" folder will have many files. One of which is "genome_complete.txt.gz" which is a compressed file of the entire aligned genome for Abusir JK2888 etc 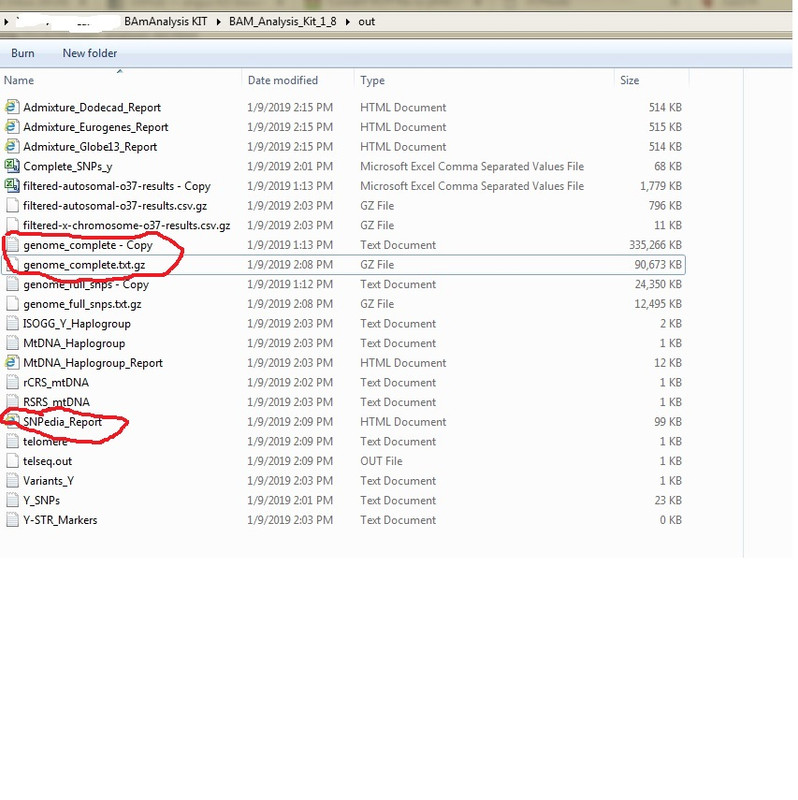  |
|
|
|
Post by djoser-xyyman on Jan 9, 2019 15:17:53 GMT -5
SNP report for ABusir jk2888
rs11803731
Location: 1:152083325
Your Genotype: AA
Summary: straighter hair (more ..)
rs763317
Location: 7:55095197
Your Genotype: AA
Summary: 3.5x increased lung cancer risk for never-smoking females (more ..)
rs7027989
Location: 9:21817754
Your Genotype: AA
Summary: 1.26x increased myocardial infarction risk (more ..)
rs10811661
Location: 9:22134094
Your Genotype: TT
Summary: 1.2x increased risk for type-2 diabetes (more ..)
rs505922
Location: 9:136149229
Your Genotype: TT
Summary: blood type O (more ..)
rs10993994
Location: 10:51549496
Your Genotype: TT
Summary: 1.2x prostate cancer risk (more ..)
rs11636232
Location: 15:28386626
Your Genotype: CC
Summary: darker eye color (more ..)
rs1426654
Location: 15:48426484
Your Genotype: AA
Summary: probably light-skinned, European ancestry (more ..)xyyman comment- KENYANS!! Henn et al
rs736839
Location: 18:46528065
Your Genotype: CC
Summary: common, increased risk for sickle cell leg ulcers (more ..)
rs6036025
Location: 20:22039868
Your Genotype: GG
Summary: more likely to go bald (more ..)
|
|
|
|
Post by djoser-xyyman on Jan 16, 2019 15:39:59 GMT -5
Ok. As I said, I update tutorial when I have something new.
So for newbies and others interested. I got PLINK2 to work.! YES!! This a major breakthrough. Why?
Plink2 can run under Windows and since people on here uses Windows they can start playing around with doing their own testing of aDNA like with the Abusir.
So we have two major pieces(Software) that run off Windows. BamAnalysis kit and PLINK2.
Procedure?
1. Download 3 Abusir bam files.
2. Download and install BAMAnalyisis kit
3. Download PLINK2. Extract. This is line command so no "installing" is necessary.
4. Botht BAmAnalysis Kit and PLINK2 runs in the DOS/CMD environment under Windows.. Unlike many other programs that run under Unix which most people are not familiar with.
5. Process the ABusir mummies using BamA Kit and get the VCF files. You can combine the VCF files or test separately for each mummy.
6. Convert the Abusir VCF files to plink files using PLINK2 to get a bim, bed and fam files
7. Use Merge/Combine plink Files with HapMAp or your test comparison dataset. There should be only one large file with all your populations including Abusirs. Use merge?catenate file command.
8. Run these files(*.bim, *.bed, *.fam) through ADMIXTURE
9. Run these ADMIXTURE results files through gplot in R to see ADMIXTURE Chart
I am still working on tagging, labeling and titling.
Working on other things also like IBD etc. KING
More to come.
|
|
|
|
Post by djoser-xyyman on Jan 16, 2019 15:40:18 GMT -5
Script used for PLINK2 eg plink2 --make-bed --vcf bam_chr3.vcf --out binary_testBYour input file and output files are in italic and could be anything for name. Specify you VCF input file and give you output file any name you wish but just remember it..especially if your working folder is filled. PLINK2 will spit out 4 files. Three will the bim, bed and fam files to be used in ADMIXTURE. 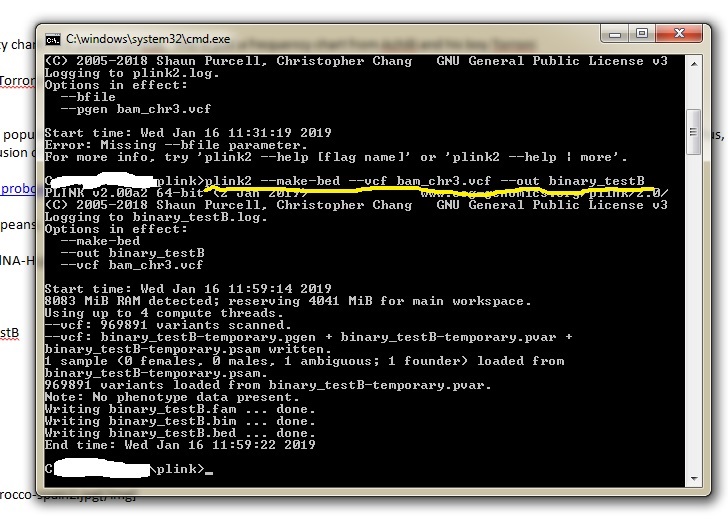 |
|
|
|
Post by djoser-xyyman on Jan 22, 2019 9:22:50 GMT -5
Fasta/fastq/BAM files to VCF files then index or sort/concat with VCFtools or bcftools.
Concatenate All Vcf.Gz Files
(assumes that your files are tabix indexed).
vcf-concat *.vcf.gz | gzip -c > out.vcf.gz
or use bcftools
-----------------------
How to concatenate “by chromosome”-VCFs?
Quote:
I have a several VCFs which are VCF which only contain information by chromosome. That is, there's a chromosome 1 VCF (with only chr1), a chromosome 2 VCF (with only chr2), etc.
I checked to make sure that these VCFs were valid via VCFtools, i.e.. vcf-validator
Quote:
I would recommend bcftools concat. You can't just cat them together because each file has a header section. The bcftools command will handle all that for you. Each vcf file must be sorted prior to calling concat
bcftools concat -o total_chroms.vcf chr1.vcf chr2.vcf chr3.vcf ... chrX.vcf
-o is the output file name
Indexing:
bcftools index -t or -c in.vcf.gz
-t=tabix -c = csi
|
|
|
|
Post by djoser-xyyman on Jan 22, 2019 11:07:18 GMT -5
vcftools [ --vcf FILE | --gzvcf FILE | --bcf FILE] [ --out OUTPUT PREFIX ] [ FILTERING OPTIONS ] [ OUTPUT OPTIONS ]
input
--vcf <input_filename>
--gzvcf <input_filename>
Filtering
--chr <chromosome>
--not-chr <chromosome>
--from-bp <integer>
--to-bp <integer>
--bed <filename>
OUTPUT OTHER FORMATS/b]
--BEAGLE-GL
--BEAGLE-PL
--plink
--plink-tped
--chrom-map
Converting VCF files to PLINK format
vcftools --vcf input_data.vcf --plink --chr 1 --out output_in_plink
|
|
|
|
Post by djoser-xyyman on Jan 22, 2019 12:38:54 GMT -5
jk2134_chr1.vcf.gz
vcf-validator jk2134_chr1.vcf.gz
vcf-sort jk2134_chr1.vcf.gz | gzip -c > sort-1-jk2134_chr1.vcf.gz
vcf-sort jk2134_chr2.vcf.gz | gzip -c > sort-1-jk2134_chr2.vcf.gz
vcf-sort jk2134_chr1.vcf.gz | gzip > sort-2-jk2134_chr1.vcf.gz
vcf-sort jk2134_chr2.vcf.gz | gzip > sort-2-jk2134_chr2.vcf.gz
vcf-concat
vcf-concat sort-1-jk2134_chr1.vcf.gz sort-1-jk2134_chr2.vcf.gz | gzip -c > combined-1-2-jk2134-jan-22-out.vcf.gz
|
|
|
|
Post by djoser-xyyman on Jan 22, 2019 12:39:52 GMT -5
next...bcftools
----
Fasta/fastq/BAM files to VCF files then index or sort/concat with VCFtools or bcftools.
Concatenate All Vcf.Gz Files
(assumes that your files are tabix indexed).
vcf-concat *.vcf.gz | gzip -c > out.vcf.gz
or use bcftools
-----------------------
How to concatenate “by chromosome”-VCFs?
Quote:
I have a several VCFs which are VCF which only contain information by chromosome. That is, there's a chromosome 1 VCF (with only chr1), a chromosome 2 VCF (with only chr2), etc.
I checked to make sure that these VCFs were valid via VCFtools, i.e.. vcf-validator
Quote:
I would recommend bcftools concat. You can't just cat them together because each file has a header section. The bcftools command will handle all that for you. Each vcf file must be sorted prior to calling concat
bcftools concat -o total_chroms.vcf chr1.vcf chr2.vcf chr3.vcf ... chrX.vcf
-o is the output file name
Indexing:
bcftools index -t or -c in.vcf.gz
-t=tabix -c = csi
|
|
|
|
Post by djoser-xyyman on Jan 23, 2019 16:21:26 GMT -5
test
freebayes -f hg19.fa chr1.bam > jk2888-chr1.vcf
Only about 14,000 autosomal SNPs were used when compared to the reference(hg19). Can someone verify?
------------------------------------------
Hg19 Reference (run in the same folder to obtain reference and index)
Just download it from here:
Step1
hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
Step2
tar -zxvf chromFa.tar.gz
step3
cat chr*.fa > hg19.fa
Done!
Take about 20min total
>>>>>>>>>>>
Index the reference with BWA
Script:
./bwa index hg19.fa
Takes about 90minutes depending on system configuration
>>>>>>>>>>>>>>>>>>>>
Running freebayes to obtain VCF file to be used with plink1.9/plink2
freebayes -f hg19.fa JK2888-bam_complete_sorted.bam > jk2888-var1.vcf
Took about 3 hrs
-------------------------------
|
|
|
|
Post by djoser-xyyman on Jan 24, 2019 21:45:54 GMT -5
freebayes -f hg19.fa JK2911udg.fixedRG.bam > jk2911-freebayes-var1.vcf
grep '^#' jk2888_chr1.vcf > jk2888_merge.vcf
grep -v '^#' jk2888_chr1.vcf jk2888_chr2.vcf jk2888_chr3.vcf jk2888_chr4.vcf jk2888_chr5.vcf jk2888_chr6.vcf jk2888_chr7.vcf jk2888_chr8.vcf jk2888_chr9.vcf jk2888_chr10.vcf jk2888_chr11.vcf jk2888_chr12.vcf jk2888_chr13.vcf jk2888_chr14.vcf jk2888_chr15.vcf jk2888_chr16.vcf jk2888_chr17.vcf jk2888_chr18.vcf jk2888_chr19.vcf jk2888_chr20.vcf jk2888_chr21.vcf >> jk2888_merge.vcf
plink --make-bed --vcf jk2134_merge.vcf --out jk2134-forplink-out
error correction
plink --make-bed --allow-extra-chr --vcf jk2134_merge.vcf --out jk2134-forplink-out
plink --make-bed --allow-extra-chr --vcf jk2911_merge.vcf --out jk2911-forplink-out
plink --make-bed --allow-extra-chr --vcf jk2888_merge.vcf --out jk2888-forplink-out
|
|
|
|
Post by djoser-xyyman on Jan 25, 2019 15:51:44 GMT -5
BTW @ newbies. I came across some NEW tools to plot ADMIXTURE . It is really simple. Now. There are several ways to convert the Abusir bam/fastq files to plink format to plot in ADMIXTURE. We can do the same with these dataset on the ancient Iberians ....WITHOUT African autosomes but African uniparental haplogroups. Wink! wink!
The easier process I just came across
1. Get the Bam/Fastq files of the ancient individuals say the Iberians above..OP
2. Download and install the following software. These are all done on a LINUX/UNIX/UBUNTU System - Freebayes, Plink19/Plink2, BWA, ADMIXTURE, R, and the human refrence-hg19 in fasta format.
3. Index the hg19 using BWA to setup you reference aligner using hg19 fasta file
4. Verify the Abusir sample sets(x3) are in bam format and in the same folder as the aligned hg19 packet(Indexed).
5. Create you Abusir VCf files using Freebayes. Eg script
freebayes -f hg19.fa JK2888udg.fixedRG.bam > jk2888-from-full-bam.vcf
6. This will kick out the Abusir file(jk2888-from-full-bam.vcf) to run through the next software. ...PLINK1.9
7. Using PLINK to create your Abusir plink files (bim,bed, fam) of the 3 abusir. One at a time.
8. Merge the 3 Abusir samples from PLINK with you database files also in PLINK format. HGDP or whichever you want to use.
9. With the new merged file run it through ADMIXTURE to get you Q file
10 Running your Q file with R software to get your barplot.
I am still working on labelling. More to come.
BTW running to freebayes can take up to 8 hours or more on the larger Abusir mummy file.
I will post pictures and a more detailed tutorial when I have the time. But it is all there above. There are other methods but so far I found this is the cleanest and fastest using Freebayes and BWA.
Installing the programs is a bear.
Oh! No.5 is simply stating "Freebayes to take file jk2888udg.fixedRG.bam, align it to the hg19 reference and kick out the VCf version of the abusir file" ...which can be used in PLINK which in turn can be used in ADMIXTURE. SIMPLE!!!!
Your file naming convention is up to you.
|
|
|
|
Post by Tukuler al~Takruri on Jan 27, 2019 21:01:10 GMT -5
One guy didn't take well to my reduxes. Hoping you find something useful about it. Sorted by • 100% frequencies • majority frequencies • plurality frequencies • tandem K's • minority I got 5 or 6, likely geographic, major populations out of the 10 allowed bins. I think 21's what they use for global analysis now. In a quick glance the redux indicates isolates, 'partners', infusions, and 'trickles'. As seen, the algorithm determines the layout. Independent minded blacks will have to design these kinds of programs but based on algorithms they will build. Using TheSet's algorithms will give you TheSet's refined data. Refined data influenced by unconscious bias, good bad indifferent, of the coder. 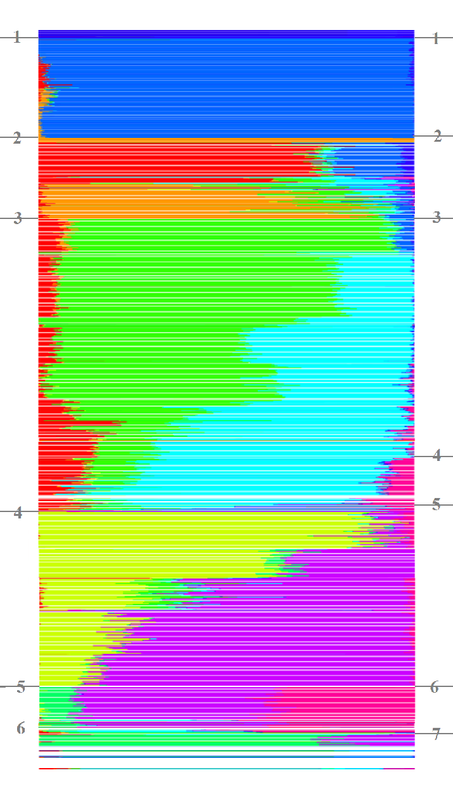 Finally first created ADMIXTURE bar plot. population chosen at k=10. Need to put labels on x-axis........but making progress ---- Grep "AfricanBarbados\|EsanNigeria\|EthiopianJews\|Gambian\|Ethiopians\|Yoruba\|Mbuti_Pygmies\|San\|Mandenka\|Bantu_NE\|Luhya\|Biaka_Pygmies\|Bedouin\|Egyptans\|Cypriots\|Druze\|Palesti nian\|AshkenazyJews\|Saudis\|SephardicJews\|Syrians\|Moroccans\|Mozabite\|Mende\|LibyaJew\|TunisiaJew\|Yemenese\|Tuscan\|Sardinian\|Sicily\|Greek\|Spaniard\|IranJew\|IraqiJews\|Ita lyJew\|Jordanians\|Sindhi\|Samaritians\|Bengali\|Gond\|Makrani\|Papuan\|NAN_Melanesian\|UtahWhite\|Finn\|French_Basque\|GreatBritain\|Mongol\|HanBeijing\|Han_S\|Dai\|Vietnamese\|Ka ritiana\|Maya\|MumbaiJews\|Peruvian" Est1000HGDP.fam > main-sample-set.txt 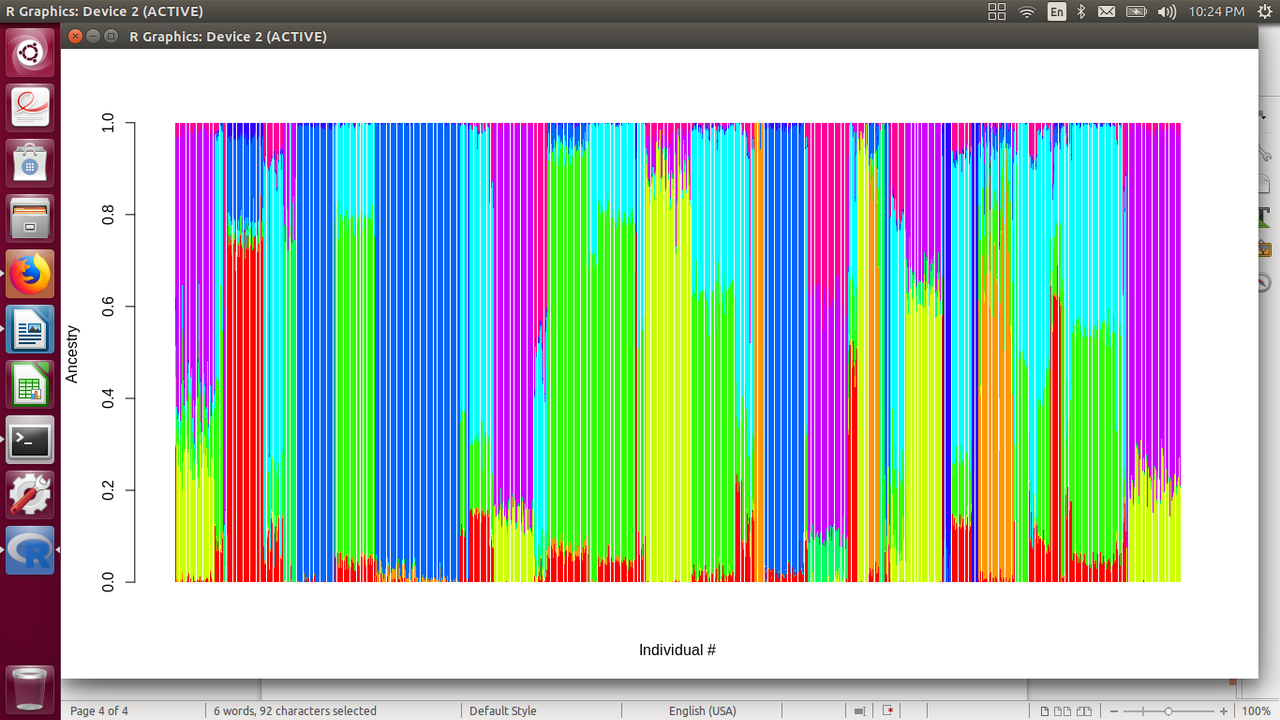 |
|
|
|
Post by djoser-xyyman on Jan 30, 2019 11:26:11 GMT -5
Ta! Still have to figure out how to keep pops together... One guy didn't take well to my reduxes. Hoping you find something useful about it. Sorted by • 100% frequencies • majority frequencies • plurality frequencies • tandem K's • minority I got 5 or 6, likely geographic, major populations out of the 10 allowed bins. I think 21's what they use for global analysis now. In a quick glance the redux indicates isolates, 'partners', infusions, and 'trickles'. As seen, the algorithm determines the layout. Independent minded blacks will have to design these kinds of programs but based on algorithms they will build. Using TheSet's algorithms will give you TheSet's refined data. Refined data influenced by unconscious bias, good bad indifferent, of the coder. Finally first created ADMIXTURE bar plot. population chosen at k=10. Need to put labels on x-axis........but making progress ---- Grep "AfricanBarbados\|EsanNigeria\|EthiopianJews\|Gambian\|Ethiopians\|Yoruba\|Mbuti_Pygmies\|San\|Mandenka\|Bantu_NE\|Luhya\|Biaka_Pygmies\|Bedouin\|Egyptans\|Cypriots\|Druze\|Palesti nian\|AshkenazyJews\|Saudis\|SephardicJews\|Syrians\|Moroccans\|Mozabite\|Mende\|LibyaJew\|TunisiaJew\|Yemenese\|Tuscan\|Sardinian\|Sicily\|Greek\|Spaniard\|IranJew\|IraqiJews\|Ita lyJew\|Jordanians\|Sindhi\|Samaritians\|Bengali\|Gond\|Makrani\|Papuan\|NAN_Melanesian\|UtahWhite\|Finn\|French_Basque\|GreatBritain\|Mongol\|HanBeijing\|Han_S\|Dai\|Vietnamese\|Ka ritiana\|Maya\|MumbaiJews\|Peruvian" Est1000HGDP.fam > main-sample-set.txt |
|
|
|
Post by djoser-xyyman on Jan 30, 2019 11:27:07 GMT -5
the latest placeholder - creating bim bed fam files for the Abusirs After creating you vcf file for each Abusir above using freebayes or other methods/script. Then... e.g. a.---- Validate it!!! vcf-validator jk2888-hg19.vcf, etc b. - ---create a tabix index for each to merge bgzip -c jk2888-hg19.vcf > jk2888-hg19.gz tabix -p vcf jk2888-hg19.gz c. ---merge using vcftools vcf-merge jk2911-hg19.gz jk2888-hg19.gz jk2134-hg19.gz | bgzip -c > jk2888-jk-2134-jk-2911-out.vcf.gz d. ---unzip and run through plink plink --make-bed --vcf jk2888-jk-2134-jk-2911-out.vcf --out jk2888-jk-2134-jk-2911-out result output- jk2888-jk-2134-jk-2911-out.bim jk2888-jk-2134-jk-2911-out.bam jk2888-jk-2134-jk-2911-out.fam These can now be run in ADMIXTURE and R 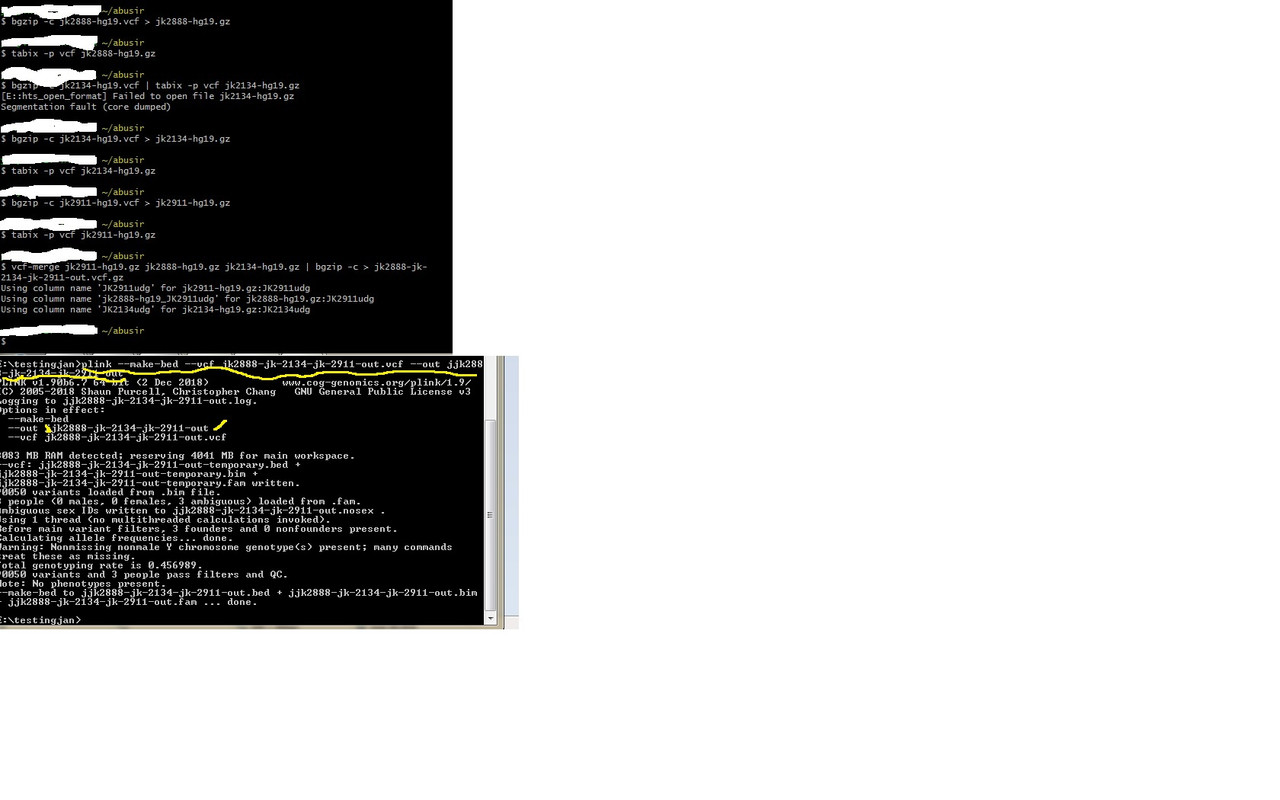 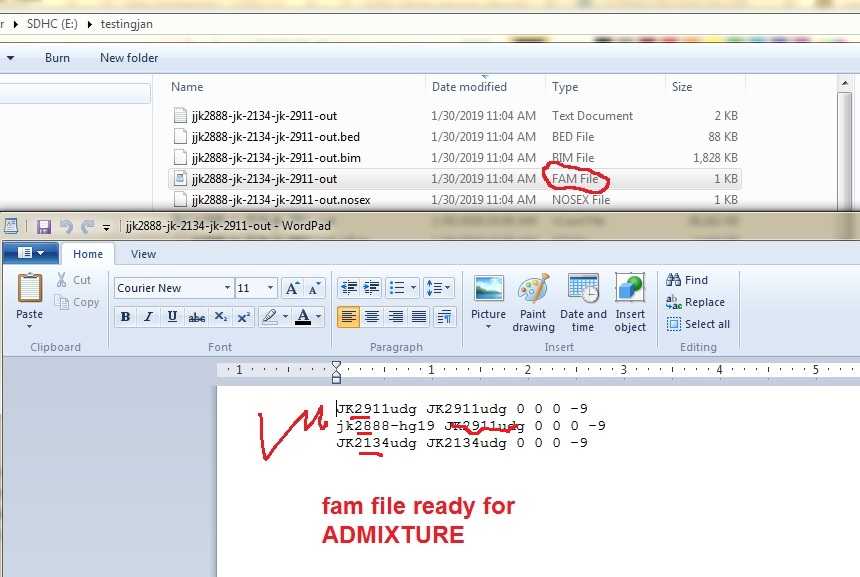 |
|



