|
|
Post by djoser-xyyman on Jul 18, 2019 13:05:01 GMT -5
I haven't read these as yet ...too busy . But they got my attention.
I have always said that Iberia and even the western coast of Africa played an important part in early human migration OOA. Even Sicily and Sardinia. It makes geographic sense. The lies about the "near East" is just that....lies and misdirection. When I get the time I will review and post, but here they are
====
Ancestral mitochondrial N lineage from the Neolithic 'green'Sahara
S Vai, S Sarno, M Lari, D Luiselli, G Manzi, M Gallinaro… - Scientific reports, 2019
Because Africa's climate hampers DNA preservation, knowledge of its genetic
variability is mainly restricted to modern samples, even though population genetics
dynamics and back-migrations from Eurasia may have modified haplotype …
Twitter Facebook
Genome-wide characterization of Arabian Peninsula populations: shedding light on the history of a fundamental bridge between continents
V Fernandes, N Brucato, JC Ferreira, N Pedro… - Molecular biology and …, 2019
Abstract The Arabian Peninsula (AP) was an important crossroad between Africa,
Asia, and Europe, being the cradle of the structure defining these main human
population groups, and a continuing path for their admixture. The screening of …
Twitter Facebook
[PDF] A western route of prehistoric human migration from Africa into the Iberian Peninsula
G González-Fortes, F Tassi, E Trucchi, K Henneberger… - Proceedings of the Royal …, 2019
Being at the western fringe of Europe, Iberia had a peculiar prehistory and a complex
pattern of Neolithization. A few studies, all based on modern populations, reported
the presence of DNA of likely African origin in this region, generally concluding it was …
Twitter Facebook
[PDF] Mitochondrial Genome Diversity in the Central Siberian Plateau with Particular Reference to Prehistory of Northernmost Eurasia
SV Dryomov, AM Nazhmidenova, EB Starikovskaya… - bioRxiv, 2019
The Central Siberian Plateau was last geographic area in Eurasia to become
habitable by modern humans after the Last Glacial Maximum (LGM). Through
comprehensive mitochondrial DNA genomes retained in indigenous Siberian …
Twitter Facebook
Have Europeans Always Arrived from the Near East?
G Vogl - Adventure Diffusion, 2019
In 2015, a widespread group of geneticists (Mathieson et al. 2015) reported the first
ancient DNA investigation of the full genome from 26 Anatolian skeletons,
concluding that these belonged to a population that was the source of Europe's first …
Twitter Facebook
[HTML] Genetic diversity of the Thao people of Taiwan using Y-chromosome, mitochondrial DNA and HLA gene systems
JA Trejaut, F Muyard, YH Lai, LR Chen, ZS Chen… - BMC evolutionary biology, 2019
Despite attempts in retracing the history of the Thao people in Taiwan using folktales,
linguistics, physical anthropology, and ethnic studies, their history remains
incomplete. The heritage of Thao has been associated with the Pazeh Western …
Twitter Facebook
[HTML] Compound-specific radiocarbon dating and mitochondrial DNA analysis of the Pleistocene hominin from Salkhit Mongolia
T Devièse, D Massilani, S Yi, D Comeskey, S Nagel… - Nature communications, 2019
A skullcap found in the Salkhit Valley in northeast Mongolia is, to our knowledge, the
only Pleistocene hominin fossil found in the country. It was initially described as an
individual with possible archaic affinities, but its ancestry has been debated since the …
Twitter Facebook
A Companion to Anthropological Genetics
DH O'Rourke - 2019
Explore the latest research in anthropological genetics and understand the genome's
role in cultural and social development A Companion to Anthropological Genetics
illustrates the role of genetic analysis in advancing the modern study of human …
Twitter Facebook
Amanda VanSteelandt and
AC Stone - A Companion to Anthropological Genetics, 2019
With mutually selective influence on growth, reproduction, and survival, pathogens
and their host organisms are engaged in an evolutionary race to evade or attack the
other. The history of human–pathogen interactions is written into the genomes of …
Twitter Facebook
[PDF] the Clock
MP Cox - A Companion to Anthropological Genetics, 2019
The role that molecular evidence can play in assigning dates to past events was first
identified in the early 1960s with the discovery of the molecular clock. Zuckerkandl
and Pauling (1962) proposed that lineages acquire mutations in a regular fashion …
|
|
|
|
Post by djoser-xyyman on Jul 18, 2019 13:30:43 GMT -5
With Europeans it is not what they say but more importantly what they DON'T say. Notice they leave out Africa when it is staring any logical person in the face that Neolithics are radiating from the SOUTH but they dont look to the south, Africa. eg It is impossible for Neolithic technology to appear in Iberia at the same time as Greece or Levant. The pattern is consistent with Isolation by distance. This is what you are seeing and the liars spin and spin and spin and spin. journals.plos.org/plosone/article?id=10.1371/journal.pone.0051106 |
|
|
|
Post by djoser-xyyman on Jul 19, 2019 22:00:39 GMT -5
Evidence of Austronesian Genetic Lineages in East Africa and South Arabia: Complex Dispersal from Madagascar and Southeast Asia
Nicolas Brucato, Veronica Fernandes, Pradiptajati Kusuma, Viktor Černý, Connie J Mulligan, Pedro Soares, T
Abstract
The Austronesian dispersal across the Indonesian Ocean to Madagascar and the Comoros has been well documented, but in an unexplained anomaly, few to no traces have been found of the Austronesian expansion in East Africa or the Arabian Peninsula. To revisit this peculiarity, we surveyed the Western Indian Ocean rim populations to identify potential Austronesian genetic ancestry. We generated full mitochondrial DNA genomes and genome-wide genotyping data for these individuals and compared them with the Banjar, the Indonesian source population of the westward Austronesian dispersal. We find strong support for Asian genetic contributions to maternal lineages and autosomal variation in modern day Somalia and Yemen. Surprisingly, this input reveals two apparently different geographic origins and timings of admixture for the Austronesian contact; one at a very early phase (likely associated with the early Austronesian dispersals), and a later movement dating to the end of nineteenth century. These Austronesian gene flows come, respectively, from Madagascar and directly from an unidentified location in Island Southeast Asia. This result reveals a far more complex dynamic of Austronesian dispersals through the Western Indian Ocean than has previously been understood and suggests that Austronesian movements within the Indian Ocean may have been part of a lengthy process, probably continuing well into the modern era.
|
|
|
|
Post by djoser-xyyman on Jul 19, 2019 22:08:14 GMT -5
Random Subspace Projection for Predicting Biogeographical Ancestry
Publisher: IEEE
4
Author(s)
Tanjin Toma ; Tayo Olufemi-Ajayi ; Jeremy Dawson ; Donald Adjeroh
Abstract:
Human biogeographical ancestry estimation using genomic information is an important problem with applications in population stratification, admixture mapping, forensic ancestry inference, and in healthcare. Various studies have proposed panels of ancestry informative single nucleotide polymorphisms (SNPs) for distinguishing between widely separated continental populations. There has been limited investigation on identifying SNP panels for sub-continental ancestry prediction, especially given the difficult challenge of identifying SNP markers to distinguish closely associated sub-populations, for instance, within a continent. In this study, we propose an ancestry informative SNP selection algorithm exploiting the concept of random subspace projection using supervised learning. The proposed approach identifies small panels of useful SNPs for subcontinental level ancestry classification. We show results for sub-continental level classification for all five continents in our dataset.
|
|
|
|
Post by djoser-xyyman on Jul 20, 2019 6:55:14 GMT -5
The Geography of Jewish Ethnogenesis
Aram Yardumian
Department of History and Social Sciences, Bryn Athyn College, Bryn Athyn, PA 19009-0717,
Theodore G. Schurr
Department of Anthropology, University of Pennsylvania, Philadelphia, PA 19104-6398, USA
Abstract
A reevaluation of the anthropological genetics literature on Jewish populations reveals them not simply to be a body of genetically related people descending from a small group of common ancestors, but rather a “mosaic” of peoples of diverse origins. Greek and other pre-medieval historiographic sources suggest the patterning evident in recent genetic studies could be explained by a major contribution from Greco-Roman and Anatolian-Byzantine converts who affiliated themselves with some iteration of Judaism beginning in the first and second centuries ce and continuing into the Middle Ages. These populations, along with Babylonian and Alexandrian Jewish communities, indigenous North Africans, and Slavic-speaking converts to Judaism, support a mosaic geography of Jewish ancestry in Europe and Western Asia, rather than one arising from a limited set of lineages originating solely in Palestine.
|
|
|
|
Post by djoser-xyyman on Jul 20, 2019 7:09:32 GMT -5
DNAxs/DNAStatistX: Development and validation of a software suite for the data management and probabilistic interpretation of DNA profiles
Author links open overlay panelCorina C.G.BenschopaJerryHoogenbooma
Abstract
The data management, interpretation and comparison of sets of DNA profiles can be complex, time-consuming and error-prone when performed manually. This, combined with the growing numbers of genetic markers in forensic identification systems calls for expert systems that can automatically compare genotyping results within (large) sets of DNA profiles and assist in profile interpretation. To that aim, we developed a user-friendly software program or DNA eXpert System that is denoted DNAxs. This software includes features to view, infer and match autosomal short tandem repeat profiles with connectivity to up and downstream software programs. Furthermore, DNAxs has imbedded the ‘DNAStatistX’ module, a statistical library that contains a probabilistic algorithm to calculate likelihood ratios (LRs). This algorithm is largely based on the source code of the quantitative probabilistic genotyping system EuroForMix [1]. The statistical library, DNAStatistX, supports parallel computing which can be delegated to a computer cluster and enables automated queuing of requested LR calculations. DNAStatistX is written in Java and is accessible separately or via DNAxs. Using true and non-contributors to DNA profiles with up to four contributors, the DNAStatistX accuracy and precision were assessed by comparing the DNAStatistX results to those of EuroForMix. Results were the same up to rare differences that could be attributed to the different optimizers used in both software programs. Implementation of dye specific detection thresholds resulted in larger likelihood values and thus a better explanation of the data used in this study. Furthermore, processing time, robustness of DNAStatistX results and the circumstances under which model validations failed were examined. Finally, guidelines for application of the software are shared as an example. The DNAxs software is future-proof as it applies a modular approach by which novel functionalities can be incorporated.
|
|
|
|
Post by djoser-xyyman on Jul 20, 2019 20:52:15 GMT -5
PAMAM: Power analysis in multiancestry admixture mappingYadu Gautam Sudhir Ghandikota Siqi Chen Tesfaye B. Mersha First published: 26 June 2019 Abstract Admixed populations arise when two or more previously isolated populations interbreed. Admixture mapping (AM) methods are used for tracing the ancestral origin of disease‐susceptibility genetic loci in the admixed population such as African American and Latinos. AM is different from genome‐wide association studies in that ancestry rather than genotypes are tracked in the association process. The power and sample size of AM primarily depend on proportion of admixture and differences in the risk allele frequencies among the ancestral populations. Ensuring sufficient power to detect the effect of ancestry on disease susceptibility is critical for interpretability and reliability of studies using AM approach. However, there is no power and sample size analysis tool existing for AM studies in admixed population. In this study, we developed power analysis of multiancestry AM (PAMAM) to estimate power and sample size for two‐way and three‐way population admixtures. PAMAM is the first web‐based bioinformatics tool developed to calculate power and sample size in admixed population under a variety of genetic and disease phenotype models. It is a valuable resource for investigators to design a cost‐efficient study and develop grant application to pursue AM studies. PAMAM is built on JavaScript back‐end with HTML front‐end. It is accessible through any modern web browser such as Firefox, Internet Explorer, and Google Chrome regardless of operating system. It is a user‐friendly tool containing links for support information including user manual and examples, and freely available at research.cchmc.org/mershalab/PAMAM/login.html.
|
|
|
|
Post by djoser-xyyman on Jul 20, 2019 20:58:13 GMT -5
Revealing the Genetic Impact of the Ottoman Occupation on Ethnic Groups of East-Central Europe and on the Roma Population of the Area
Zsolt Bánfai,
Abstract
History of East-Central Europe has been intertwined with the history of Turks in the past. A significant part of this region of Europe has been fallen under Ottoman control during the 150 years of Ottoman occupation in the 16–17th centuries. The presence of the Ottoman Empire affected this area not only culturally but also demographically. The Romani people, the largest ethnic minority of the East-Central European area, share an even more eventful past with Turkish people from the time of their migration throughout Eurasia and they were a notable ethnic group in East-Central Europe in the Ottoman era already. The relationship of Turks with East-Central European ethnic groups and with regional Roma ethnicity was investigated based on genome-wide autosomal single nucleotide polymorphism data. Population structure analysis, ancestry estimation, various formal tests of admixture and DNA segment analyses were carried out in order to shed light to the conclusion of these events on a genome-wide basis. Analyses show that the Ottoman occupation of Europe left detectable impact in the affected East-Central European area and shaped the ancestry of the Romani people as well. We estimate that the investigated European populations have an average identity-by-descent share of 0.61 with Turks, which is notable, compared to other European populations living in West and North Europe far from the affected area, and compared to the share of Sardinians, living isolated from these events. Admixture of Roma and Turks during the Ottoman rule show also high extent
|
|
|
|
Post by djoser-xyyman on Jul 20, 2019 21:03:10 GMT -5
Collective and single burial in Madagascar Mike Parker Pearson, Denis Regnier 2019 HAL Id: hal-01882328 hal.archives-ouvertes.fr/hal-01882328Submitted on 16 Nov 2018 HAL is a multi-disciplinary open access archive for the deposit and dissemination of scientific research documents, whether they are published or not. The documents may come from teaching and research institutions in France or
abroad, or from public or private research centers quote: "The view that the Austronesian-derived Malagasy brought collective burial with their migration from South-East Asia – anthropologically hypothesised rather than archaeologically demonstrated – has led scholars to infer that this practice was always present and dominant in highland Madagascar (Metcalf & Huntington 1991: 108-109)."  reading this paper it seems Madagascar is filled with stone tombs just as N. East African and parts of Southern East African |
|
|
|
Post by djoser-xyyman on Jul 20, 2019 21:10:01 GMT -5
Inference of Population Structure from Time-Series Genotype Data Tyler A. Joseph1, * and Itsik Pe’er1,2,3, * Sequencing ancient DNA can offer direct probing of population history. Yet, such data are commonly analyzed with standard tools that assume DNA samples are all contemporary. We present DyStruct, a model and inference algorithm for inferring shared ancestry from temporally sampled genotype data. DyStruct explicitly incorporates temporal dynamics by modeling individuals as mixtures of unobserved populations whose allele frequencies drift over time. We develop an efficient inference algorithm for our model using stochastic variational inference. On simulated data, we show that DyStruct outperforms the current state of the art when individuals are sampled over time. Using a dataset of 296 modern and 80 ancient samples, we demonstrate DyStruct is able to capture a well-supported admixture event of steppe ancestry into modern Europe. We further apply DyStruct to a genome-wide dataset of 2,067 modern and 262 ancient samples used to study the origin of farming in the Near East. We show that DyStruct provides new insight into population history when compared with alternate approaches, within feasible run time. github.com/tyjo/dystructDYSTRUCT |
|
|
|
Post by djoser-xyyman on Jul 21, 2019 13:18:06 GMT -5
What really happened in the Trans Atlantic Slave trade. Did it really happen. The genetic data emerging do NOT support occurrence of a Slave Trade as we were taught. ==== Ancient DNA and bioarchaeological perspectives on European and African diversity and relationships on the colonial Delaware frontier Raquel E. Fleskes1 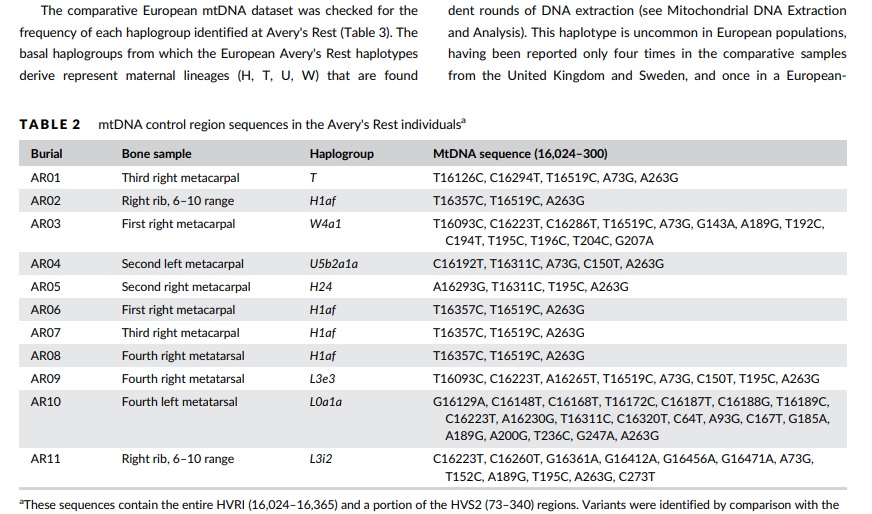 . quote: "The African haplotypes were compared to the African mtDNA dataset with respect to their frequency and geographic distribution (Tables S3–S5). Haplogroup frequency data for L0a1, L3i, and L3e were generated, as well as corresponding sub-haplotypes, as they are more numerous than the L0a1a, L3i2, and L3e3 haplotypes alone. The absence of haplogroup sharing among the three individuals of African descent indicate that they are not related maternally and possibly originated in geographically dispersed populations in Africa. It is also possible that these persons originated from the same geographic area within a genetically heterogeneous population. The larger haplogroups (L0a1 and L3e) that included the mtDNAs of the two adult males have wide distributions and occur in moderate frequencies in areas associated with 17th century slave trading ports in west and central Africa (Walsh, 2001, 2010) (Figure 5). L0a, the basal haplogroup from which L0a1 derives, was involved in the eastern Bantu expansion into the African continent, which may explain its high frequency around Mozambique (Beleza, Gusmão, Amorim, Carracedo, & Salas, 2005; Rito et al., 2013). It is absent from the
central-western coast of Nigeria, Cameroon, and Gabon. Several of the founder types of L0a leading to L0a1 are also found in Angola, suggesting some connection between the eastern and western parts of southern Africa (Beleza et al., 2005; Salas et al., 2004). Haplogroup L3e is distributed across central and southern Africa, and is also present along the western coast (Figure 5). Its derivative, L3e3, is found primarily in West Africa, although it is also present in southern and central regions (Salas et al., 2002; Soares et al., 2012). The distribution of L3e3 in the African continent likely results from persons involved in the southern Bantu agricultural expansions from West Africa carrying these mtDNAs into southwest and southern" "Specifically, L0a1a is identified in Cubans (Mendizabal et al.,
2008) and Bermudians (Gaieski et al., 2011), while L3e3 has been
reported in the Caribbean (Mendizabal et al., 2008; Salas, Carracedo,
Richards, & Macaulay, 2005) and at high frequency in African-descent
populations in Brazil (Carvalho, Bortolini, dos Santos, & Ribeiro-dosSantos, 2008; González et al., 2006; Salas et al., 2002; Silva et al., 2006). In North America, both haplogroups Le3e and L0a1 have been identified in multiple studies assaying mtDNA diversity in contemporary African American populations (Diegoli et al., 2009; Ely, Wilson, Jackson, & Jackson, 2006; Johnson et al., 2015; Salas et al., 2004). Th e L3i haplotype of the 5-year-old child (AR11) had not been
reported in African-descended populations in the Americas prior to
this study. This haplogroup is found almost exclusively in eastern
Africa (Cerezo et al., 2016; Soares et al., 2012) (Figure 5), and the" The aDNA do NOT match the account of slavery. It the scenario is more consistent with the presence of Africans prior in the Americas prior to the European arrival. |
|
|
|
Post by djoser-xyyman on Jul 21, 2019 13:29:33 GMT -5
Putting RFMix and ADMIXTURE to the test in a complex admixed population Caitlin Uren1$ , Eileen G. Hoal Abstract 27 28 Global and local ancestry inference in admixed human populations can be performed 29 using computational tools implementing distinct algorithms, such as RFMix and 30 ADMIXTURE. The accuracy of these tools has been tested largely on populations 31 with relatively straightforward admixture histories but little is known about how well 32 they perform in more complex admixture scenarios. Using simulations, we show that 33 RFMix outperforms ADMIXTURE in determining global ancestry proportions in a 34 complex 5-way admixed population. In addition, RFMix correctly assigns local 35 ancestry with an accuracy of 89%. The increase in reported local ancestry inference 36 accuracy in this population (as compared to previous studies) can largely be 37 attributed to the recent availability of large-scale genotyping data for more 38 representative reference populations. The ability of RFMix to determine global and 39 local ancestry to a high degree of accuracy, allows for more reliable population 40 structure analysis, scans for natural selection, admixture mapping and case-control 41 association studies. This study highlights the utility of the extension of computational 42 tools to become more relevant to genetically structured populations, as seen with 43 RFMix. This is particularly noteworthy as modern-day societies are becoming 44 increasingly genetically complex and some genetic tools are therefore less 45 appropriate. We therefore suggest that RFMix be used for both global and local
46 ancestry estimation in complex admixture scenarios. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry InferenceBrian K. Maples Simon Gravel Eimear E. Kenny 8 Carlos D. Bustamante Loter: A Software Package to Infer Local Ancestry for a Wide Range of Species Thomas Dias-Alves, Julien Mairal, Michael G B Blum Molecular Biology and Evolution, Volume 35, Issue 9, September 2018, Pages 2318–2326, doi.org/10.1093/molbev/msy126Published: 20 June 2018 pdfPDF Split View Cite Permissions Icon Permissions Share Abstract Admixture between populations provides opportunity to study biological adaptation and phenotypic variation. Admixture studies rely on local ancestry inference for admixed individuals, which consists of computing at each locus the number of copies that originate from ancestral source populations. Existing software packages for local ancestry inference are tuned to provide accurate results on human data and recent admixture events. Here, we introduce Loter, an open-source software package that does not require any biological parameter besides haplotype data in order to make local ancestry inference available for a wide range of species. Using simulations, we compare the performance of Loter to HAPMIX, LAMP-LD, and RFMix. HAPMIX is the only software severely impacted by imperfect haplotype reconstruction. Loter is the less impacted software by increasing admixture time when considering simulated and admixed human genotypes. For simulations of admixed Populus genotypes, Loter and LAMP-LD are robust to increasing admixture times by contrast to RFMix. When comparing length of reconstructed and true ancestry tracts, Loter and LAMP-LD provide results whose accuracy is again more robust than RFMix to increasing admixture times. We apply Loter to individuals resulting from admixture between Populus trichocarpa and Populus balsamifera and lengths of ancestry tracts indicate that admixture took place ∼100 generations ago. We expect that providing a rapid and parameter-free software for local ancestry inference will make more accessible genomic studies about admixture processes. |
|
|
|
Post by djoser-xyyman on Jul 21, 2019 18:04:18 GMT -5
Methodological issues in the Indo-European debate - Michel Danino July2019
Abstract
The Indo-European debate has been going on for a century and a half. Initially confined to linguistics, race-based anthropology and comparative mythology, it soon extended to archaeology, especially with the discovery of the Harappan civilization, and peripheral disciplines such as agriculture, archaeometallurgy or archaeoastronomy. The latest entrant in the field, archaeogenetics, is currently all but claiming that it has finally laid to rest the whole issue of a hypothetical migration of Indo-Aryan speakers to the Indian subcontinent in the second millennium BCE. This paper questions the finality of this claim by pointing to inherent limitations, methodological issues and occasional biases in current studies as well as in the interpretation of archaeological evidence.
|
|
|
|
Post by djoser-xyyman on Jul 21, 2019 18:49:11 GMT -5
Dissecting in silico Mutation Prediction of Variants in African Genomes: Challenges and Perspectives
Christian Domilongo Bope july2019
Abstract
Genomic medicine is set to drastically improve clinical care globally due to high throughput technologies which enable speedy in silico detection and analysis of clinically relevant mutations. However, the variability in the in silico prediction methods and categorization of functionally relevant genetic variants can pose specific challenges in some populations. In silico mutation prediction tools could lead to high rates of false positive/negative results, particularly in African genomes that harbor the highest genetic diversity and that are disproportionately underrepresented in public databases and reference panels. These issues are particularly relevant with the recent increase in initiatives, such as the Human Heredity and Health (H3Africa), that are generating huge amounts of genomic sequence data in the absence of policies to guide genomic researchers to return results of variants in so-called actionable genes to research participants. This report (i) provides an inventory of publicly available Whole Exome/Genome data from Africa which could help improve reference panels and explore the frequency of pathogenic variants in actionable genes and related challenges, (ii) reviews available in silico prediction mutation tools and the criteria for categorization of pathogenicity of novel variants, and (iii) proposes recommendations for analyzing pathogenic variants in African genomes for their use in research and clinical practice. In conclusion, this work proposes criteria to define mutation pathogenicity and actionability in human genetic research and clinical practice in Africa and recommends setting up an African expert panel to oversee the proposed criteria.
Keywords: African genome, incidental findings, actionable variants, whole exome sequencing, whole genome sequencing, precision medicine, pathogenicity
Table 1
Published whole exome and genomes data from sub-Saharan Africa.
|
|
|
|
Post by djoser-xyyman on Jul 24, 2019 6:43:34 GMT -5
Runs of homozygosity in sub-Saharan African populations provide insights into complex demographic histories
FC Ceballos, S Hazelhurst, M Ramsay - Human Genetics, 2019
The study of runs of homozygosity (ROH) can shed light on population demographic
history and cultural practices. We present a fine-scale ROH analysis of 1679
individuals from 28 sub-Saharan African (SSA) populations along with 1384 …
A genomic inference of the White Plymouth Rock genealogy
Y Guo, M Lillie, Y Zan, J Beranger, A Martin… - Poultry Science, 2019
Crossing of populations has been, and still is, a central component in domestication
and breed and variety formation. It is a way for breeders to utilize heterosis and to
introduce new genetic variation into existing plant and livestock populations. During …
A genetic analysis of the Gibraltar Neanderthals
L Bokelmann, M Hajdinjak, S Peyrégne, S Brace… - Proceedings of the National …, 2019
The Forbes' Quarry and Devil's Tower partial crania from Gibraltar are among the first
Neanderthal remains ever found. Here, we show that small amounts of ancient DNA
are preserved in the petrous bones of the 2 individuals despite unfavorable climatic
THE REVOLUTION OF ANCIENT DNA—WHAT DOES GENETICS TELL US ABOUT THE PAST?
Y Mathov, L Carmel
DNA is a central component in the cells of all living organisms, which contains
information that is critical for building the body and maintaining its biological
processes. Today, owing to new technological developments, scientists can
Forensic inference of biogeographical ancestry from genotype: The Genetic Ancestry Lab
D McNevin - Wiley Interdisciplinary Reviews: Forensic Science
Short tandem repeat (STR) profiling of DNA has become ubiquitous in forensic
practice and is used to associate people, objects, and places with each other and
with crimes. STRs can include or exclude a suspect or victim as the d
|
|

