|
|
Post by djoser-xyyman on Sept 16, 2018 5:49:57 GMT -5
This was generated automatically by the kit. I chose just one Chromosome and got this result for one Abusir. Apparently the Kit is an amalgam of different software which includes DodeCard v3. It Also uses LobSTR which was modified to remove CODIS autosomal DNA. See Changelog ver 1.5. So they knew what they were doing when they released the Abusir genome. The size of the files are also extremely small for a human genome granted it is aDNA and the complete genome will not be intact. I am trying out figure out how to modify the Analysis Kit to re-insert the original LobSTR program. The Kit is essentially a "batch" file....looking at the splash screen. If you a familiar with coding you will know what I am talking about. There are work arounds. We will see. Also we can run LobSTR independently but in a Unix/Linux type environment which means another PC or OS. They tried to cover their bases. Using STRaitRazor is another option. It may take me some time..... I don't know the 1st thing about it but don't you want results from filtered-autosomal-o37-results.csv.gz from the BAM Analysis Kit instead of Dienekes' dodecad admixture results? Using BAM Kit(SNP) I got this for ONE chromosome for one(1) of the 3 Abusir. There is a African presence. But we are only looking at ONE Chromosome from ONE ABusir. Trying to find out what is "Mediterranean"/ But there is no definition 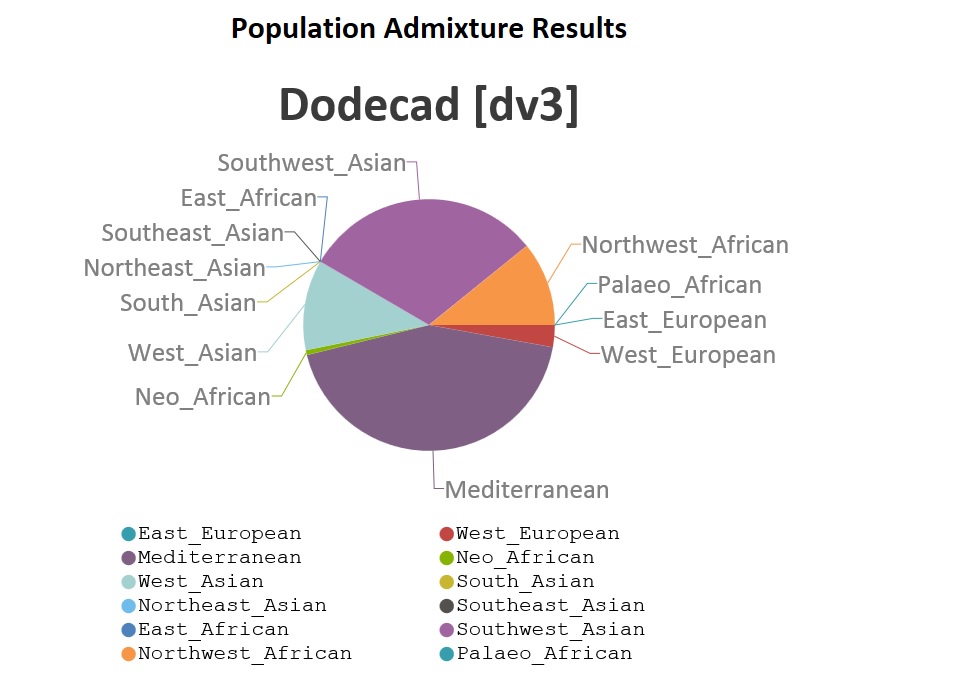 |
|
|
|
Post by djoser-xyyman on Sept 16, 2018 5:56:54 GMT -5
The quickest option is to get my hands on Kit ver 1.5 or 1.4. But these old versions were removed from Github. They know what they are doing. I am hoping someone saved the old versions. The old versions disappeared since before the flood of aDNA
|
|
|
|
Post by djoser-xyyman on Sept 18, 2018 7:32:41 GMT -5
|
|
|
|
Post by djoser-xyyman on Sept 18, 2018 7:47:39 GMT -5
Summary?
STraitRazor is Windows based program. It should be able to pull CODIS STR from GWS(autosomal DNA).
CODIS STRs is what was used to identify the Amarna(king Tut) mummies as sub-saharan Africans.
We can see that STRaitRazor can pull STRs(CODIS?)..not sure.
STRs are pulled using the software but the Excel based macro sheet should be able to ID the CODIS STRs.
The "allsequences" text file should contain the CODIS STRs which needs to be plugged into the spreadsheet which will then spit out the CODIS STRs.
|
|
|
|
Post by djoser-xyyman on Sept 18, 2018 9:40:19 GMT -5
Can confidently conclude the STR - D16S539 was REMOVED or not uploaded to the Abusir mummy XXX4488. I will assume ALL the CODIS STRs were removed. Damn shame! They know what they were doing when the made it freely available. 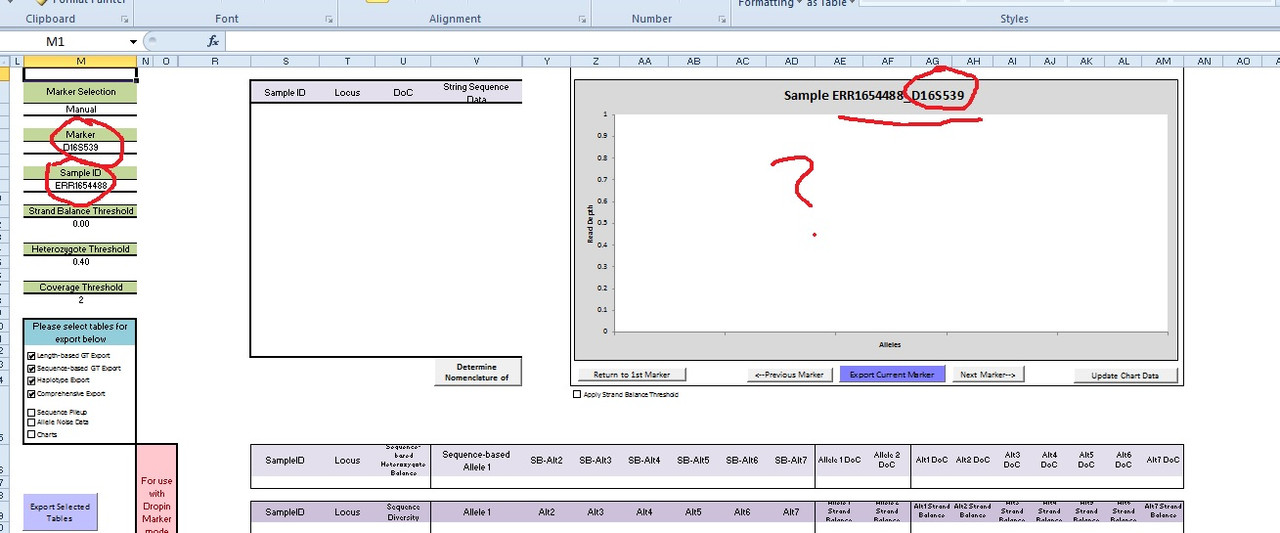 |
|
|
|
Post by djoser-xyyman on Sept 18, 2018 13:57:25 GMT -5
Using lobSTR ver 4.0.6 !!!!!! OK. So it looks like the CODIS STRs were removed from the Abusir. At least using STRaitRazor. So decdied to use another software. LobSTR version 4.0.6 This software is Linux/Unix based so not all of you may follow Anywho! Here is what I have so far. I got LobSTR running. Now it is time to play around with the 3 Abusir mummies. 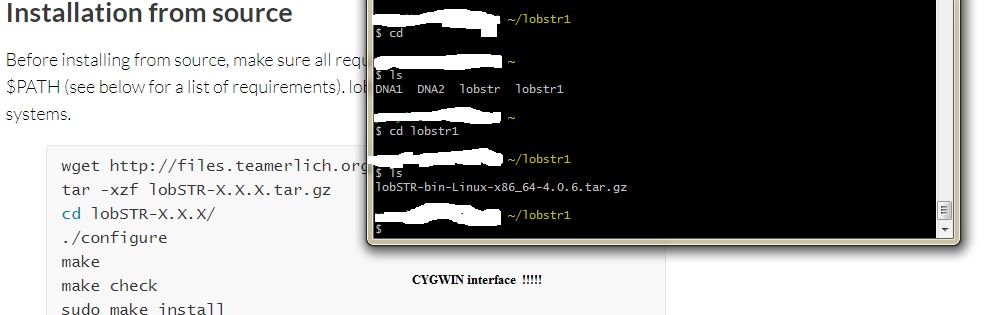 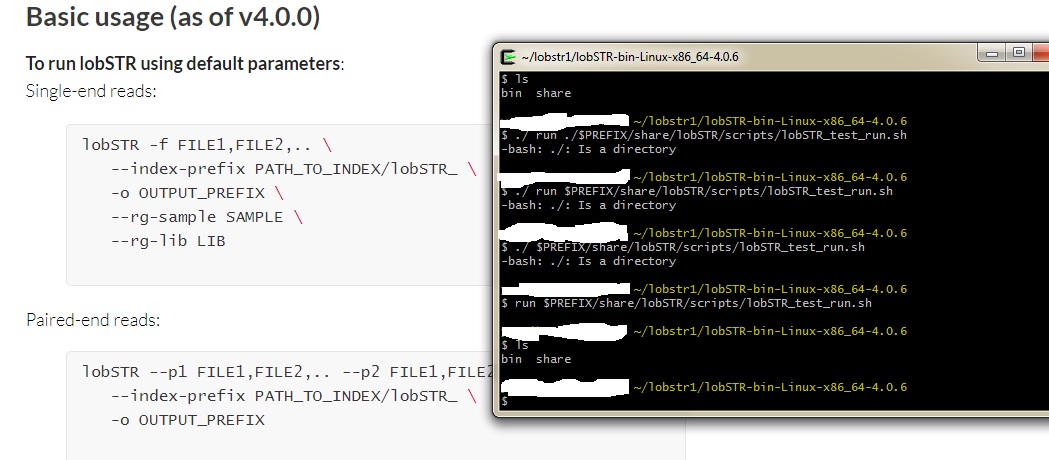  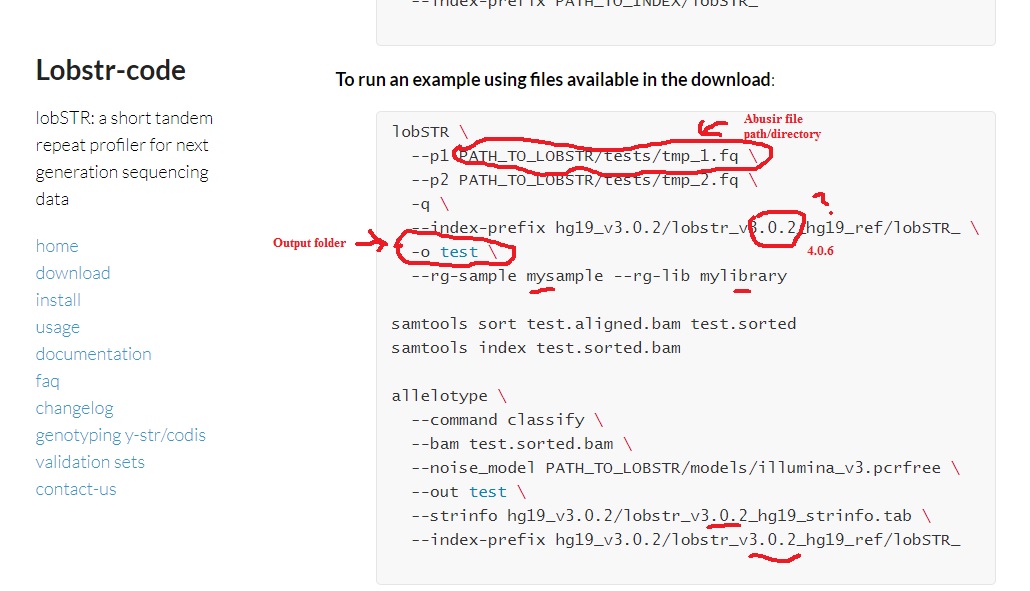 |
|
|
|
Post by djoser-xyyman on Sept 18, 2018 15:39:14 GMT -5
So it looks like this may be easier than I thought. SAMTOOLs can take a BAM files and convert to format to run on LobSTR??!! Then we can alleotype ie CODIS alignment?  ------------- 1.0 Overview One of the most common questions we receive is about how to use lobSTR to genotype Y-STRs and the CODIS set from sequencing data. Both are useful for identification in forensics settings, and Y-STRs are also used both in the genetic genealogy community and population genetics. Here we give a short tutorial on how to go from the lobSTR VCF output to a list of Y-STR and CODIS genotypes in standard nomenclature. As an example we will use the Venter genome.++ Preliminaries Before starting this tutorial, you should have: installed lobSTR (see download and install pages) downloaded the hg19 lobSTR index downloaded the Y-STR and/or CODIS marker tables. installed bedtools lobstr.teamerlich.org/best-practices-wgs-wes.html |
|
|
|
Post by djoser-xyyman on Sept 28, 2018 9:57:37 GMT -5
Abusir result from an app I used. No Bedouin connection. 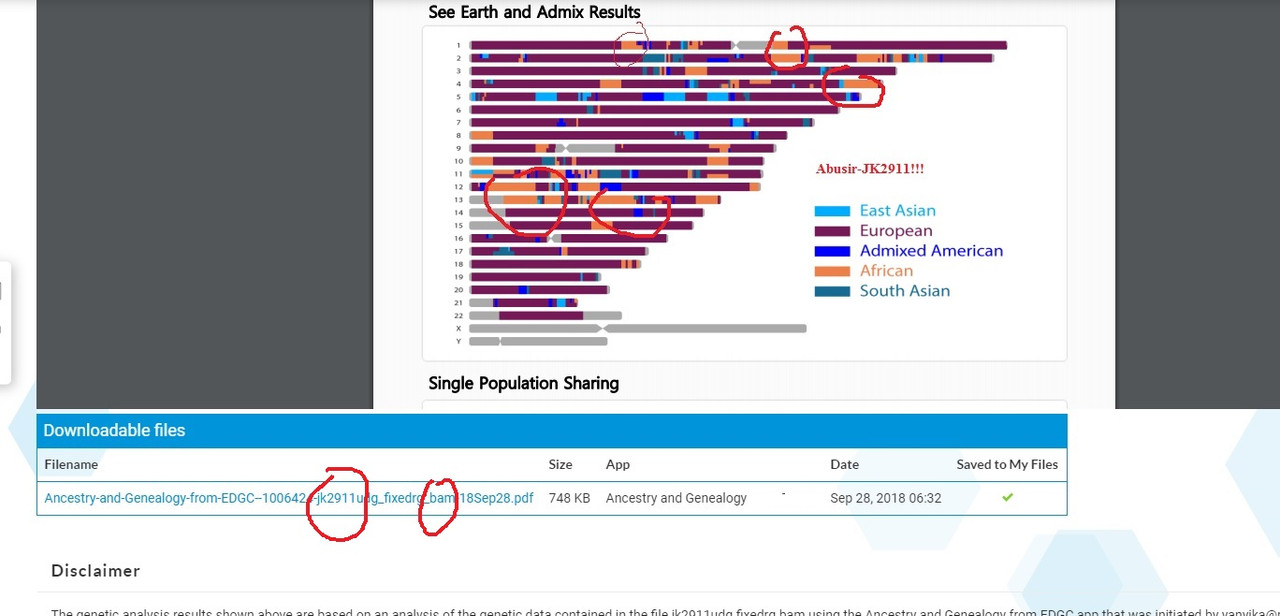 more to come |
|
|
|
Post by djoser-xyyman on Sept 28, 2018 11:36:31 GMT -5
|
|
Deleted
Deleted Member
Posts: 0
|
Post by Deleted on Sept 30, 2018 8:25:30 GMT -5
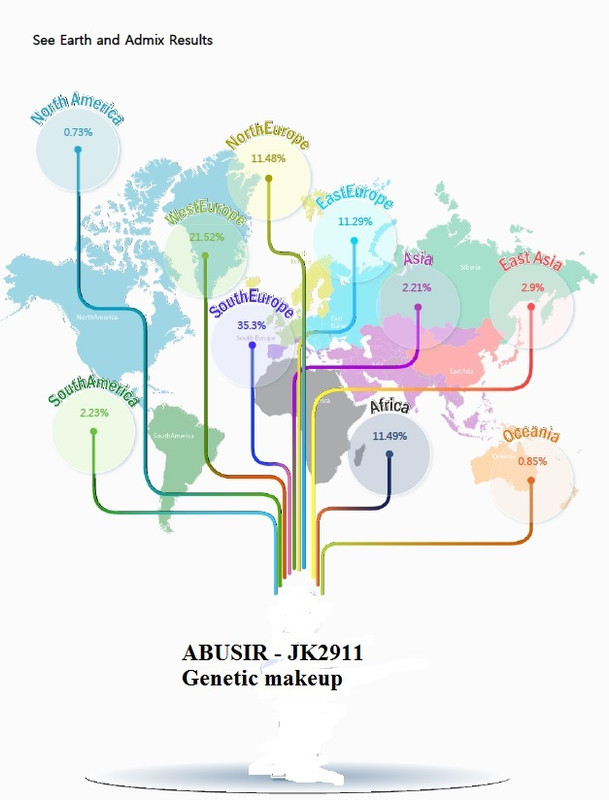
So, when all is said and done, Abusir-jk2911 turns out to be 35.3% SouthEurope, 21.52% WestEurope, 11.48% NorthEurope, 11.29% EastEurope and 11.49% Africa. Based on your analysis Abusir-jk2911 is overwhelmingly European (79.59%) and only 11.49% African. Am I reading you correctly? |
|
|
|
Post by djoser-xyyman on Oct 1, 2018 7:22:58 GMT -5
Accordng to this analysis...yes. Why did I post this? Where Jk2911 shows yup to be overwhelmingly "European"
|
|
|
|
Post by djoser-xyyman on Oct 1, 2018 7:30:35 GMT -5
Answer: It does not make sense! This online tool shows JK29111 having absolutely zero connnection with the Levant. And a strong connection with Sardinia And Iberia and a minor connection (11%) with Africa. This is why we need the CODIS STRs. Remember the original paper , Schuenemann et al, show no "African" connection prior to the Roman period. And Strong connection with Levant. While this analysis shows 11% African connection. What gives? I am now trying to run Luxmanda who has 60% Sardinian ancestry 3000BCE. It is taking a lot fo my time. Still having problem with samtools. These damn Limux/UNIX systems taking me back to my college years!!
|
|
|
|
Post by djoser-xyyman on Oct 1, 2018 10:18:32 GMT -5
This what one of the Abusir JK2911 looks like using online analysis. The "European" component is "southern" Iberia and Sardinia/Italy ie EurAfricans. Keep in mind their comparison dataset is not known. Plus CODIS STR will give a totally DIFFERENT picture. What they are saying is that this Abusir was "Southern" Europeans and NOT modern Levantines. 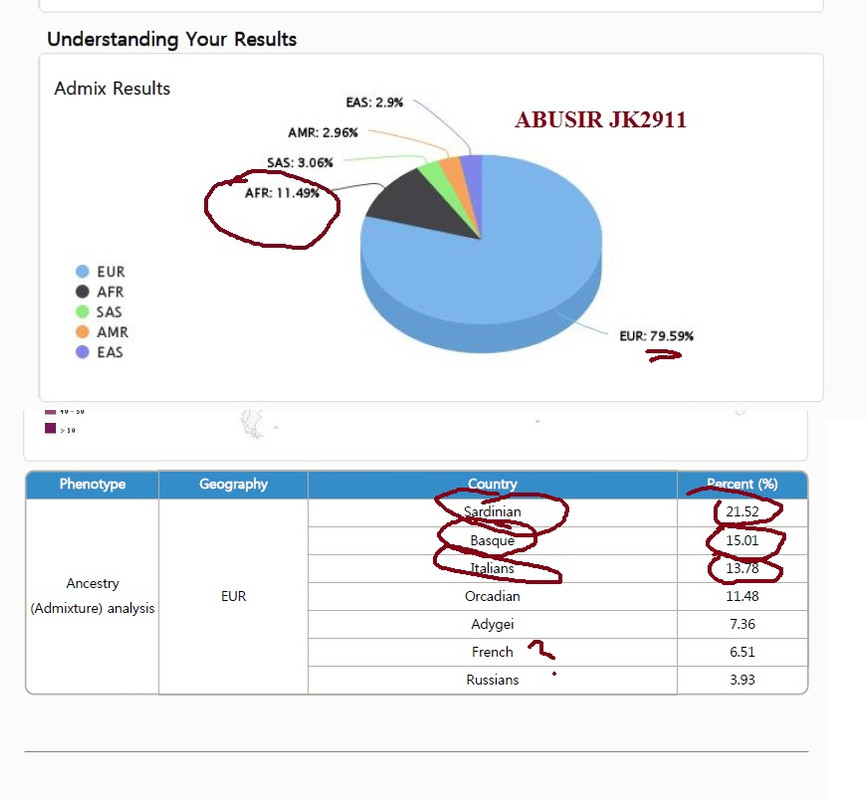 To those who are not geographically challenged and is following . We are back to the Villabruna(Sardinia) Cluster. Remember Villabruna cluster did NOT originate from the Near East/Levant according to Lazaridis2018 he is speculating either from Africa or Caucasus caves(lol!). But he confirms that Europeans did not back-migrate to the Levant. So what Are we left with? That is why CODIS STRs are so important. Why? Europeans are 80% African (Rosenberge et al 2002). The only way to differentiate Europeans from Africans is via CODIS STR? |
|
|
|
Post by djoser-xyyman on Oct 1, 2018 10:37:33 GMT -5
Now you understand the games being played and why the STRs of the Abusir were NOT released. Software like LobSTR and STRait Razor (there may be others) are showing that information can be pulled, more than what was released by the authors. I pulled many informative SNPs from the JK2911(Abusir) but I am trying to determine what it means.
|
|
|
|
Post by djoser-xyyman on Oct 4, 2018 13:53:19 GMT -5
Extracting Y-STRs and CODIS markers and converting to standard nomenclature
You can use the intersectBed tool from bedtools to extract these markers from your VCF and convert to standard notation:
lobSTR reports alleles as the number of base pairs length difference from reference (GB field of the VCF). However standardized nomenclature for these markers is given in the number of repeat units. Therefore, it is necessary to convert lobSTR results to standard nomenclature before comparing to external datasets. Y-STR and CODIS markers are included in the lobSTR reference bed file and were extracted to separate files for convenience.
Similarly for the CODIS markers, but looking at both alleles reported:
----
intersectBed -a venter.vcf -b lobSTR_codis_markers_hg19.bed -wa -wb | \
cut -f 1,2,10,14- | sed 's/:/\t/g' | cut -f 1,2,4,7,11- | sed 's/\//\t/' | \
awk '{print $0 "\t" $7+$4/$6 "\t" $7+$5/$6}'
----
Below are the results for the CODIS markers in Venter. Note, unlike the Y-STRs these can be heterozygous. We list below which alleles have evidence in the Venter reads. These will be much less accurate than the Y-STR markers since they require higher coverage to get right. Additionally, we have fewer ground truth datasets for these so there are likely to be some annotation discrepancies with standard nomenclature. If a non-unit and a unit allele were both reported and are close together, only the unit allele is kept.
|
|